Team lcolladotor Journal Club: Accurate sample deconvolution of pooled snRNA-seq using sex-dependent gene expression patterns
doi: https://doi.org/10.1093/nargab/lqaf156 (updated doi)
Presented By: Manisha Barse
Date: April 23, 2025
Recording link to our journal club meeting: https://youtu.be/zpeCFwYfV3o
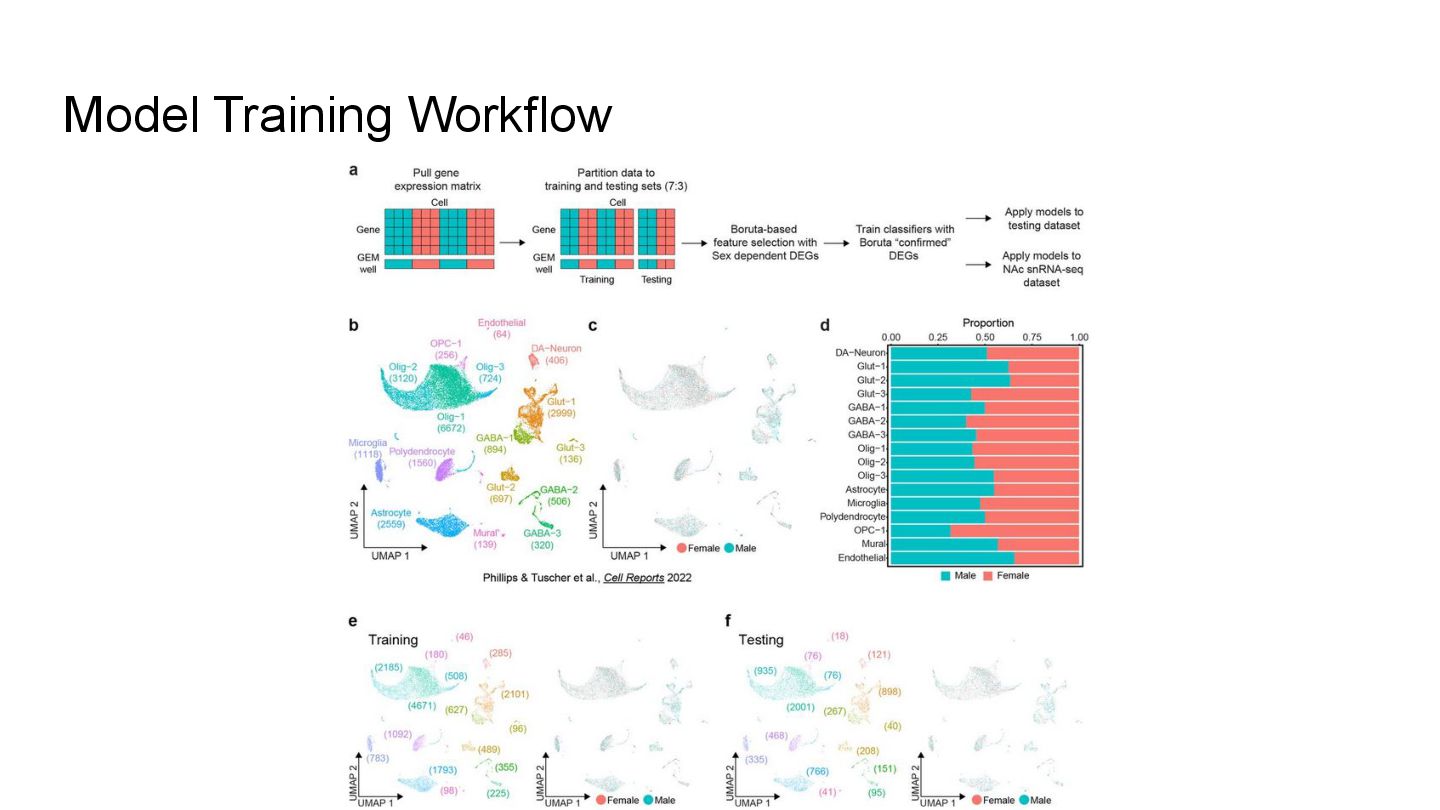
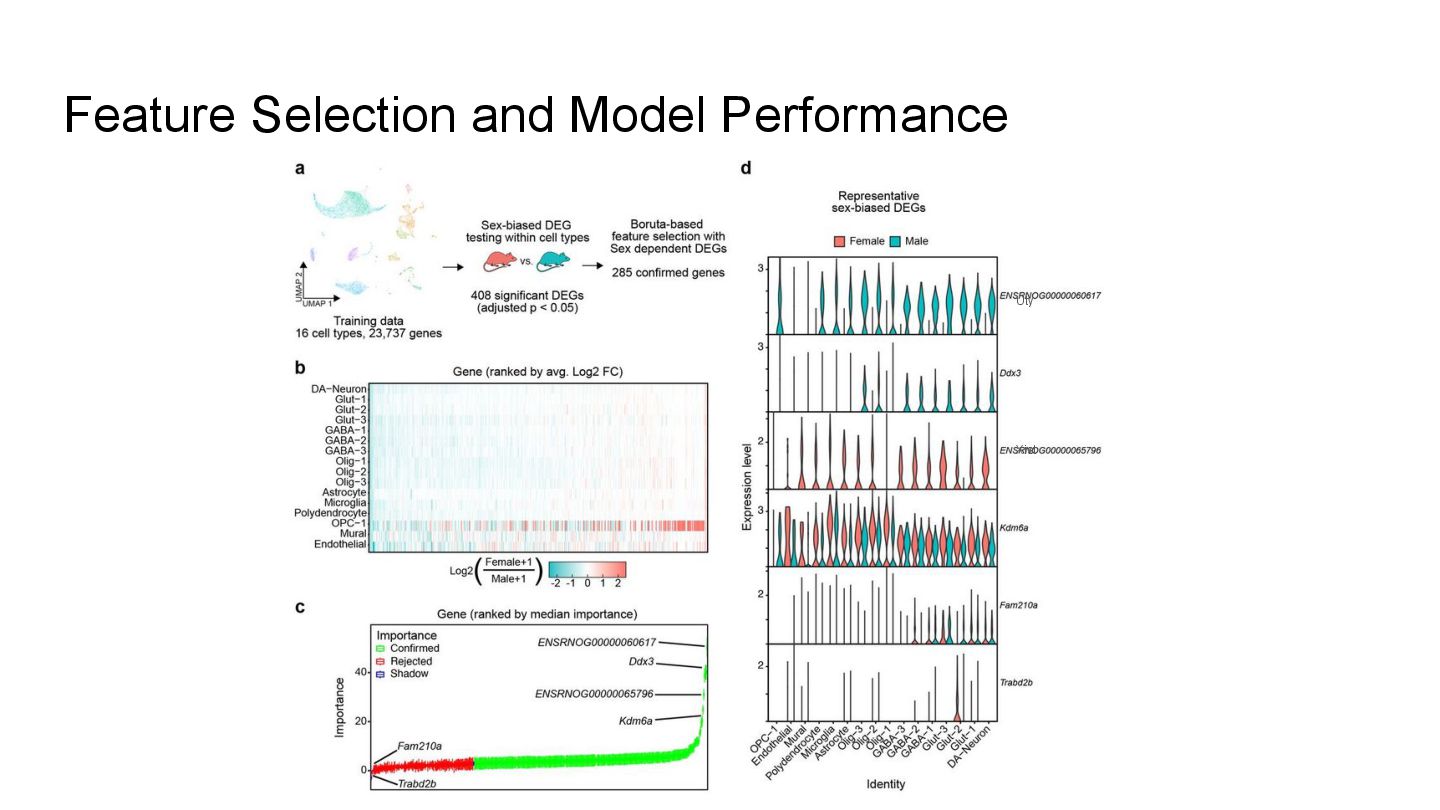
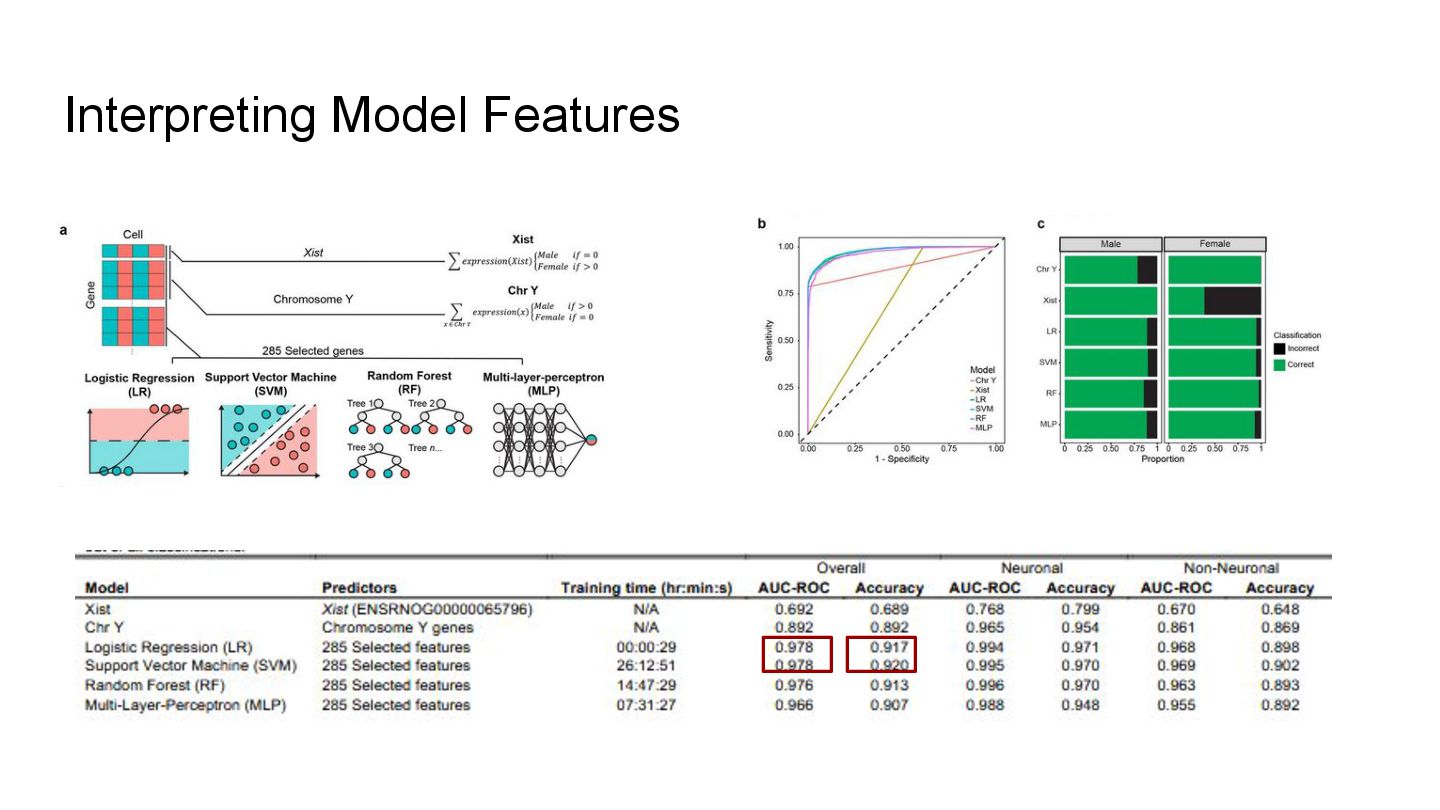
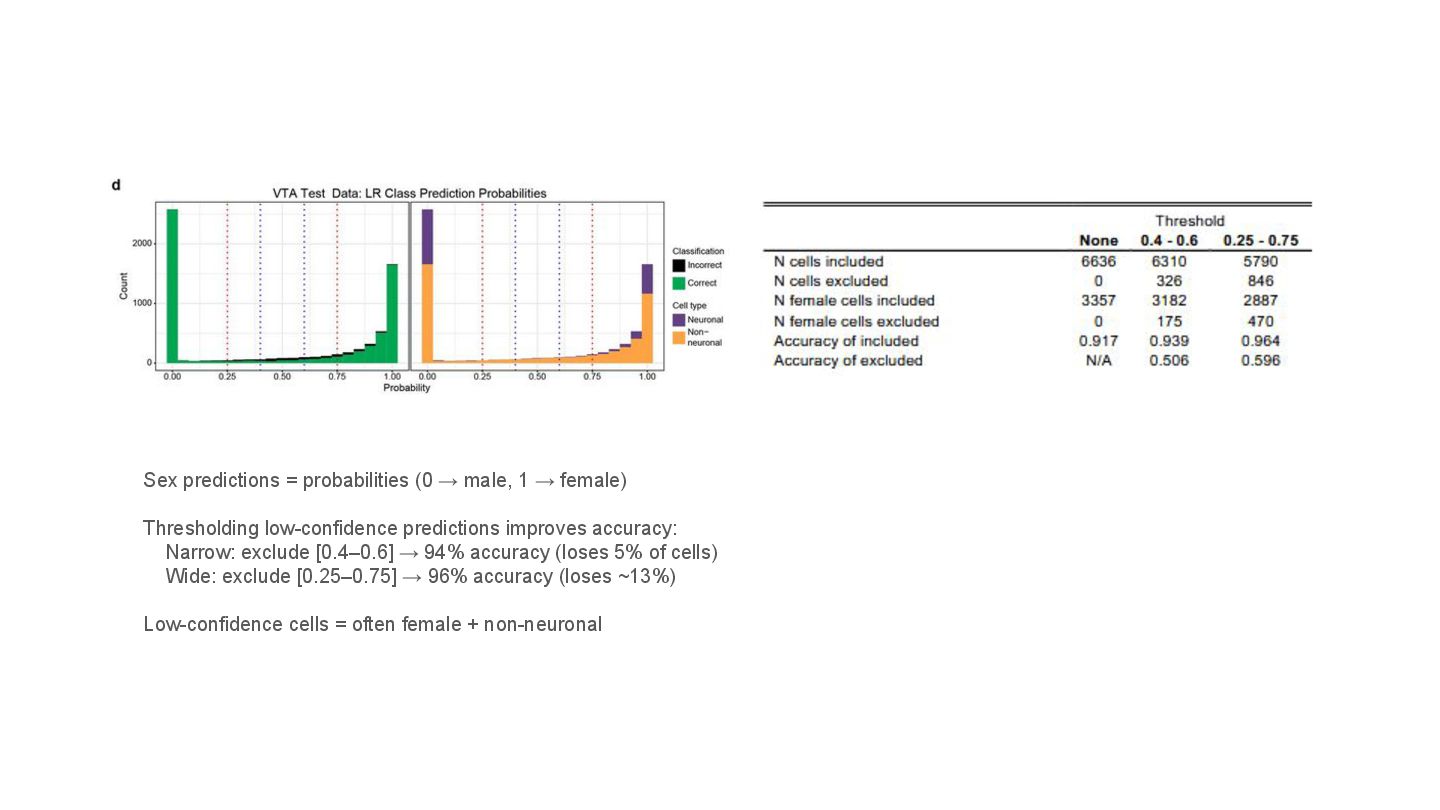
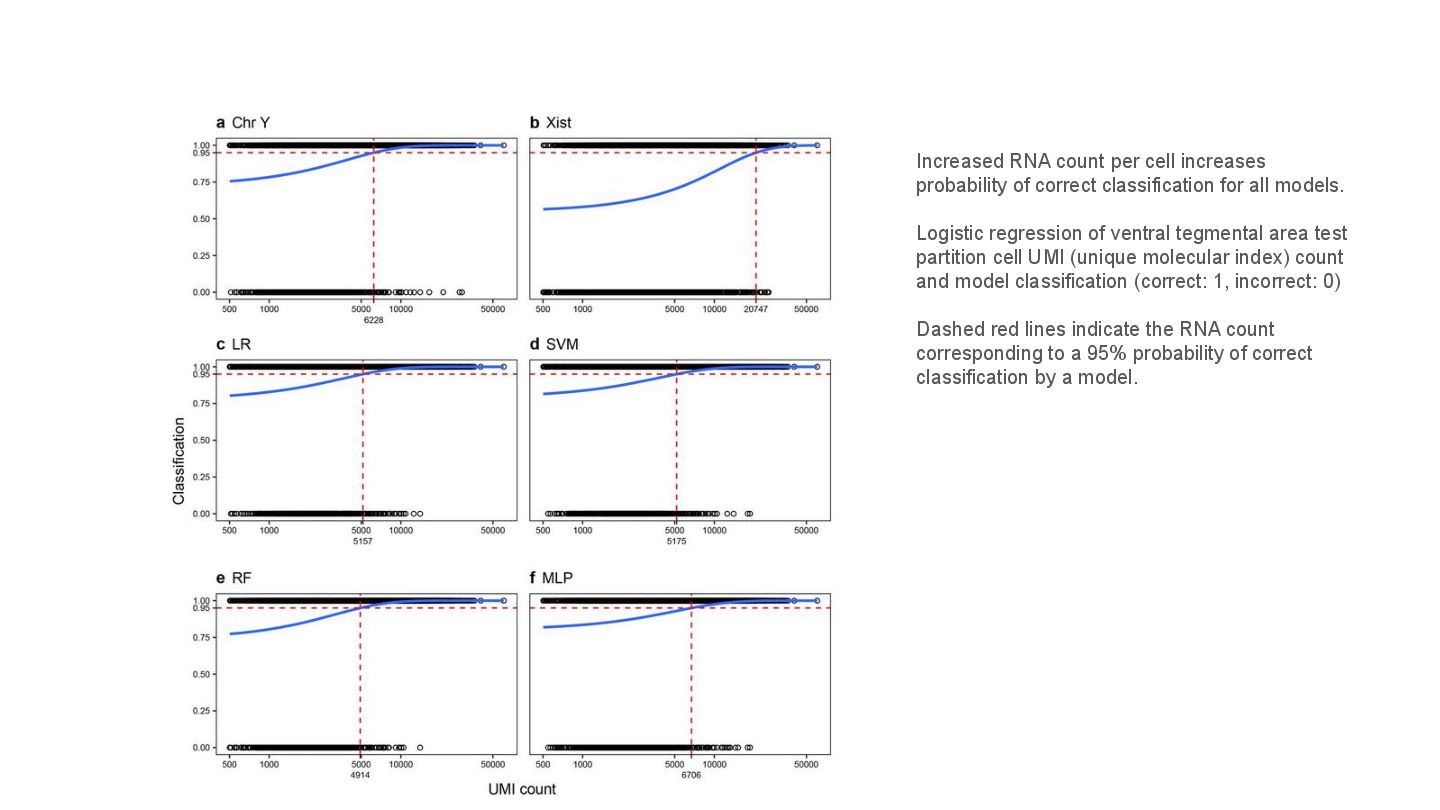
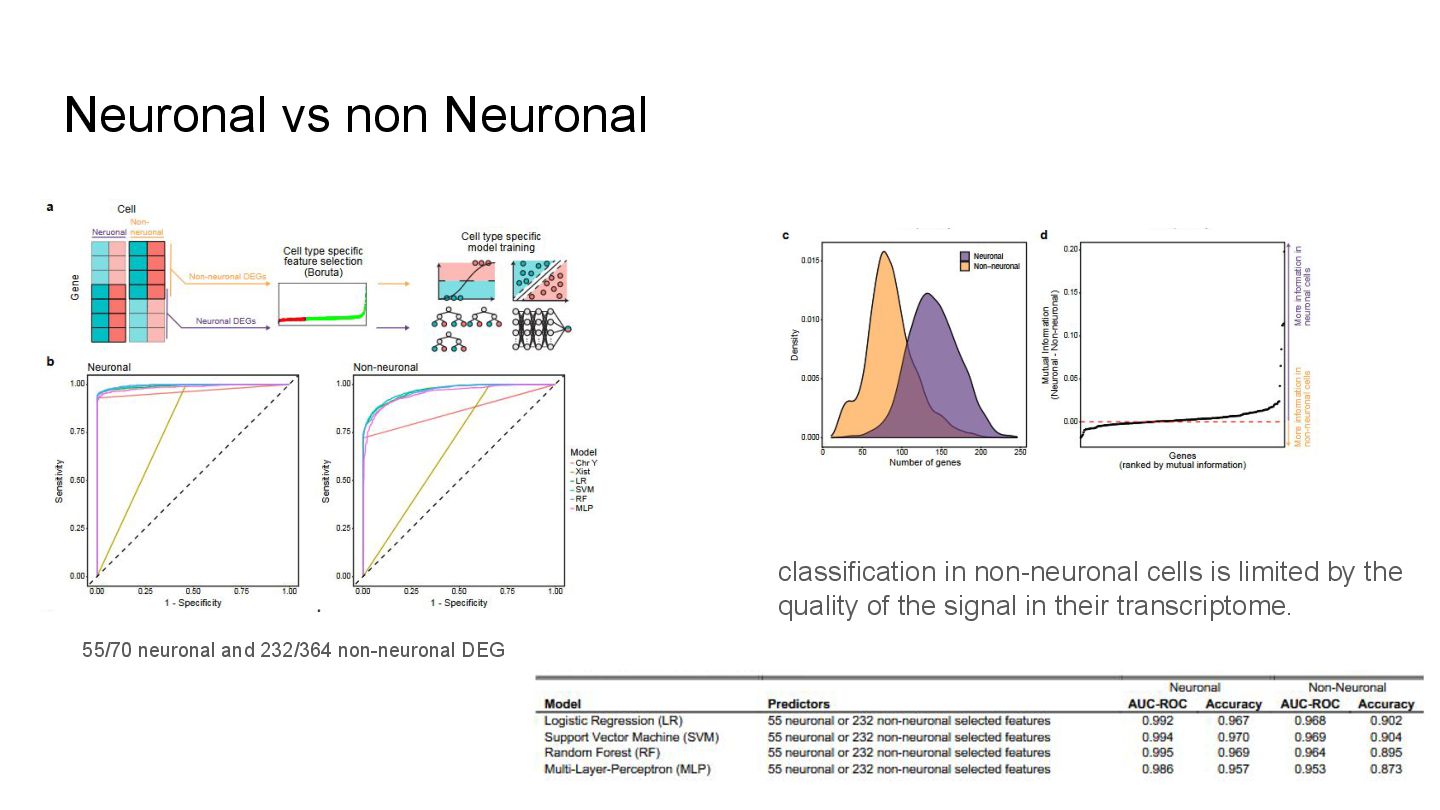
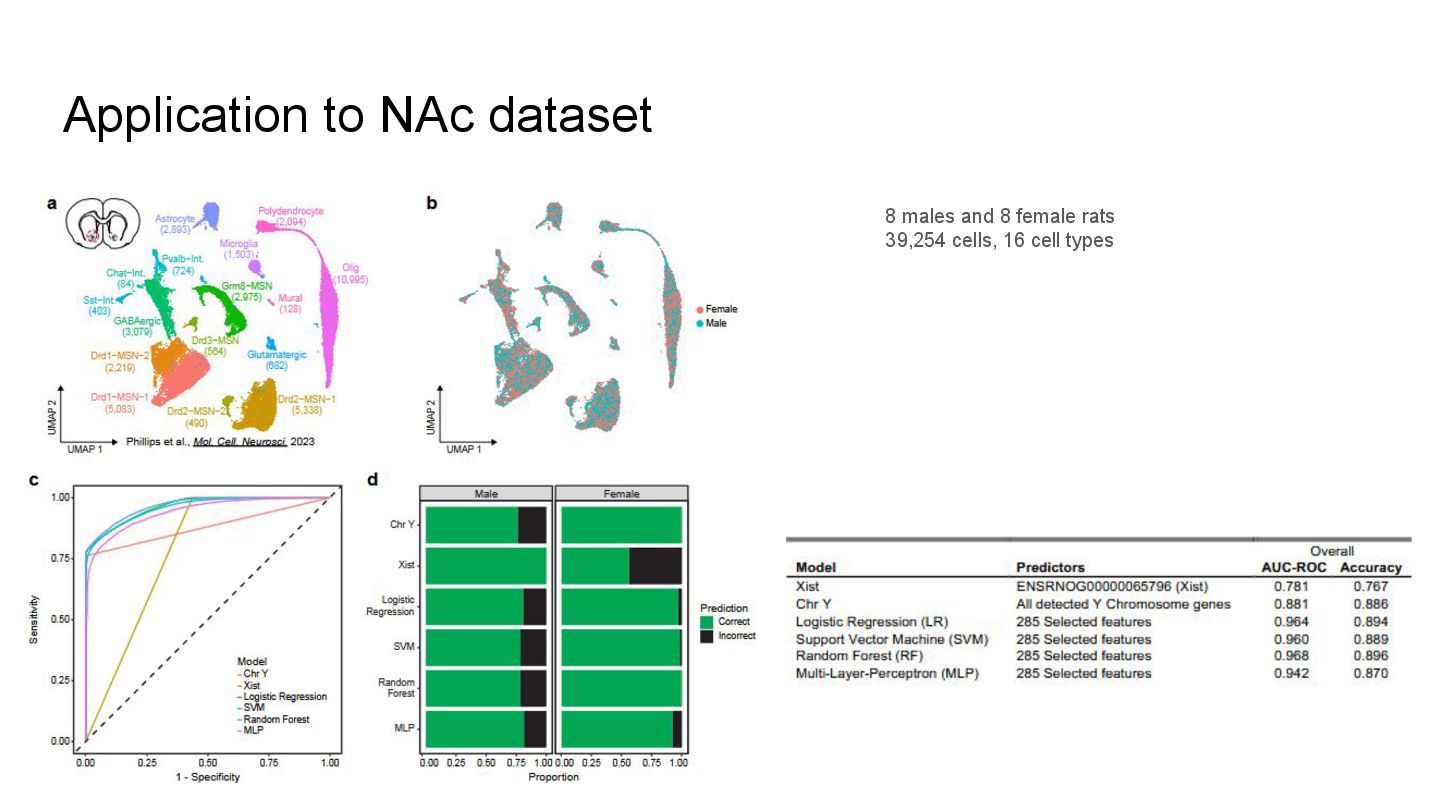
This presentation explores a machine learning approach to deconvolve pooled sn RNA-seq data by predicting cell sex, enabling cost-effective demultiplexing without experimental labels.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}