LIBD Rstats Club: pseudobulk analysis using pseudoBulkDGE()
Presented By : Manisha Barse
Date: May 02, 2025
Recording link to our rstats club presentation: https://youtu.be/0OkWBLyLrfg
GitHub Link:
https://github.com/manishabarse/LIBD_presentation/blob/main/pseudobulk_demo.R
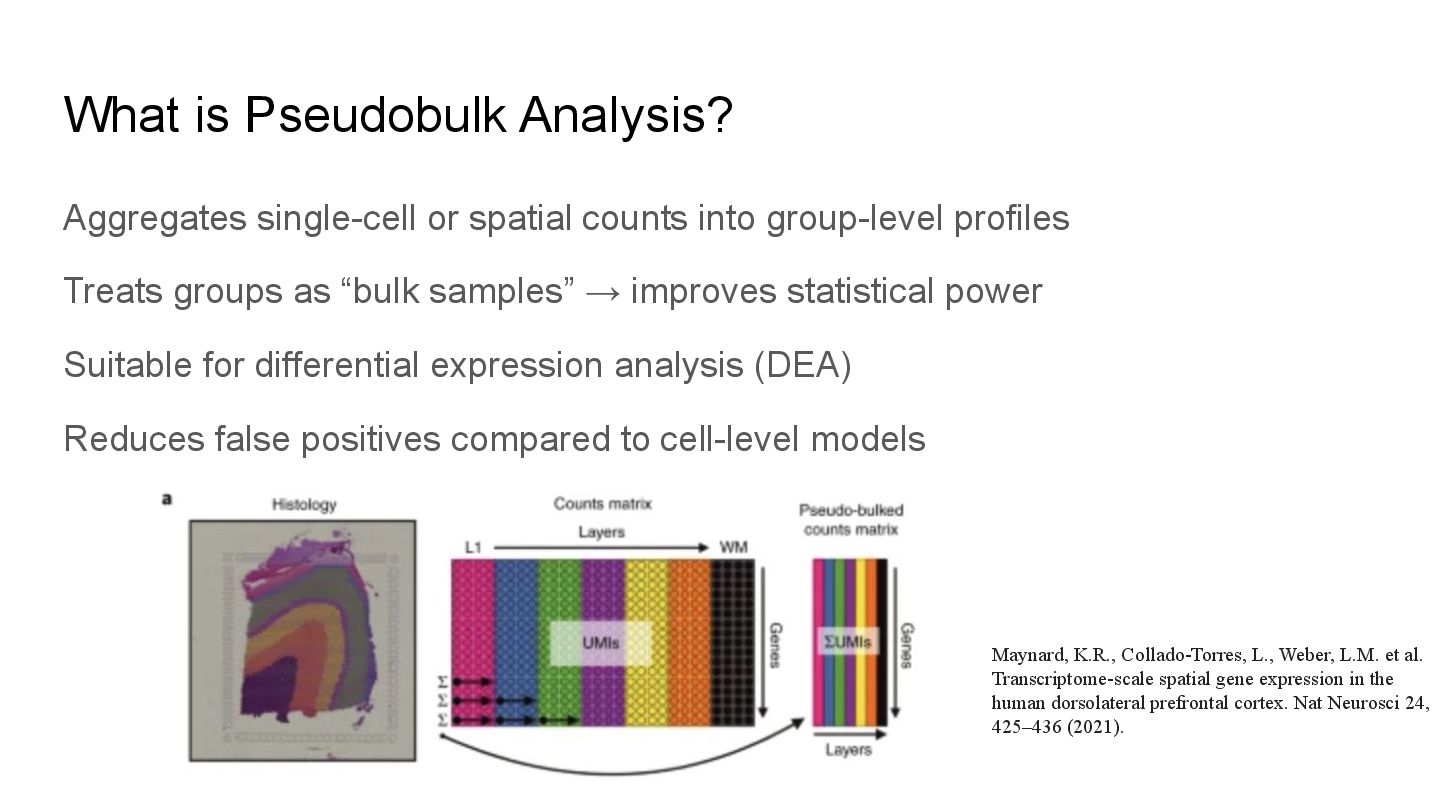
pseudoBulkDGE(): A handy wrapper for DE analysis following OSCA guidelines
#RStats #transcriptomics #Bioinformatics
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}