Adrian Schreyer Department of Biochemistry, University of Cambridge Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 1 / 46
between all molecules found in experimentally-determined biological assemblies Also contains intramolecular interactions of these molecules Contacts are represented as Structural Interaction Fingerprints (SIFts) Contains a sequence-to-structure mapping to integrate protein sequence data External resources are integrated to annotate data in CREDO Complete cheminformatics toolkits (OpenEye, RDKit) Python Application-Programming Interface (API) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 3 / 46
128,776 biological assemblies 607,505 protein-ligand interactions (not the total number of small molecules) 266,062 protein-protein interfaces, 17,793 protein-nucleic acid grooves 20 carbohydrate chains! 1,166,380,424 contacts Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 4 / 46
Structural Interaction Fingerprints (SIFts) Aromatic ring interactions Ligand-ligand interactions Data Validation Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 5 / 46
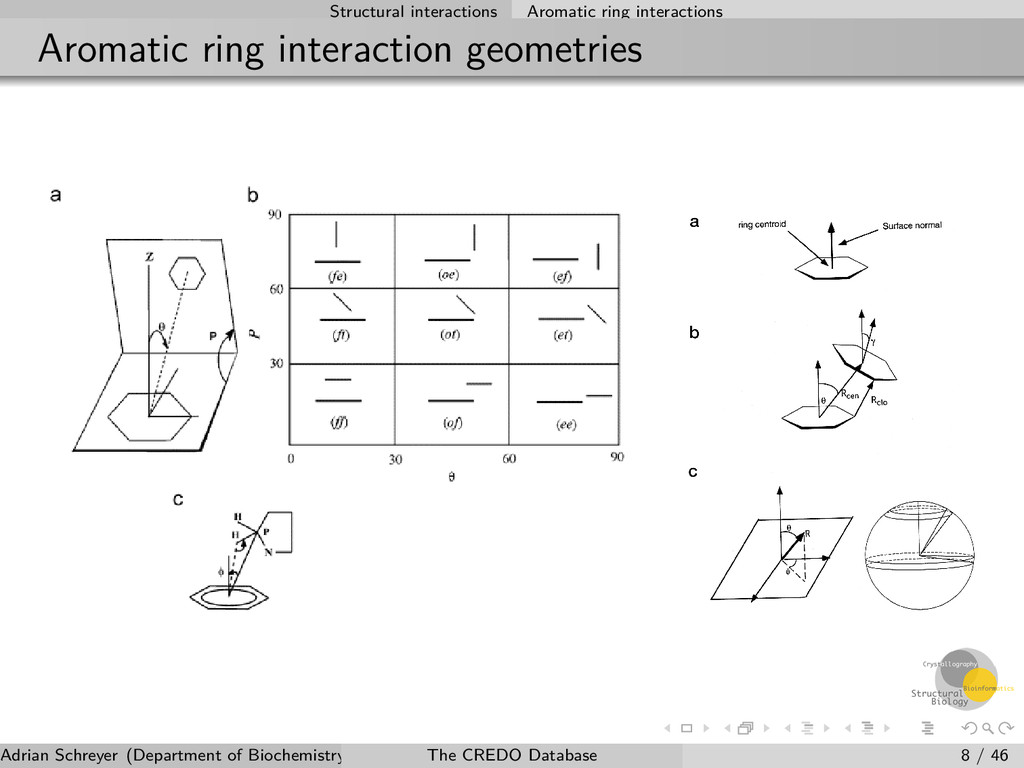
Atom and contact types Atom types are identified using SMARTS patterns Contact types are assigned based on a combination of atom types and geometrical constraints which have to be fulfilled Charges (ionisation states) are not required to determine ionic contacts Multiple contact types possible but at least one type must be present 12 interatomic interaction types 9 ring-ring interaction geometries 4 ring-atom interaction types Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 6 / 46
Interaction Fingerprints (SIFts) Aromatic ring interactions Ligand-ligand interactions Data Validation Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 7 / 46
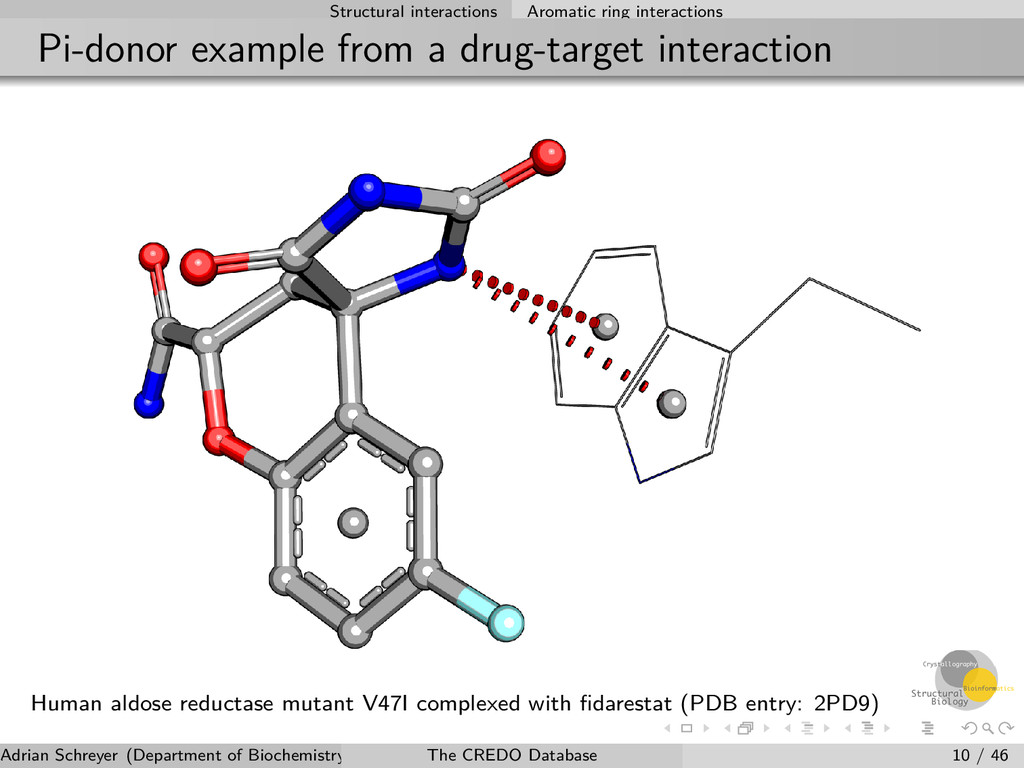
atom type Delocalised π-electron cloud of aromatic ring systems creates negative charge on both faces Can act as hydrogen bond acceptor and negatively ionisable group Distance- and geometry-dependent Interaction types π-donor: with hydrogen bond donors π-cation: with positively ionisable groups π-carbon: with weak hydrogen bond donors π-halogen: weak hydrogen bonds with halogens in a head-on orientation Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 9 / 46
interaction Human aldose reductase mutant V47I complexed with fidarestat (PDB entry: 2PD9) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 10 / 46
Fingerprints (SIFts) Aromatic ring interactions Ligand-ligand interactions Data Validation Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 11 / 46
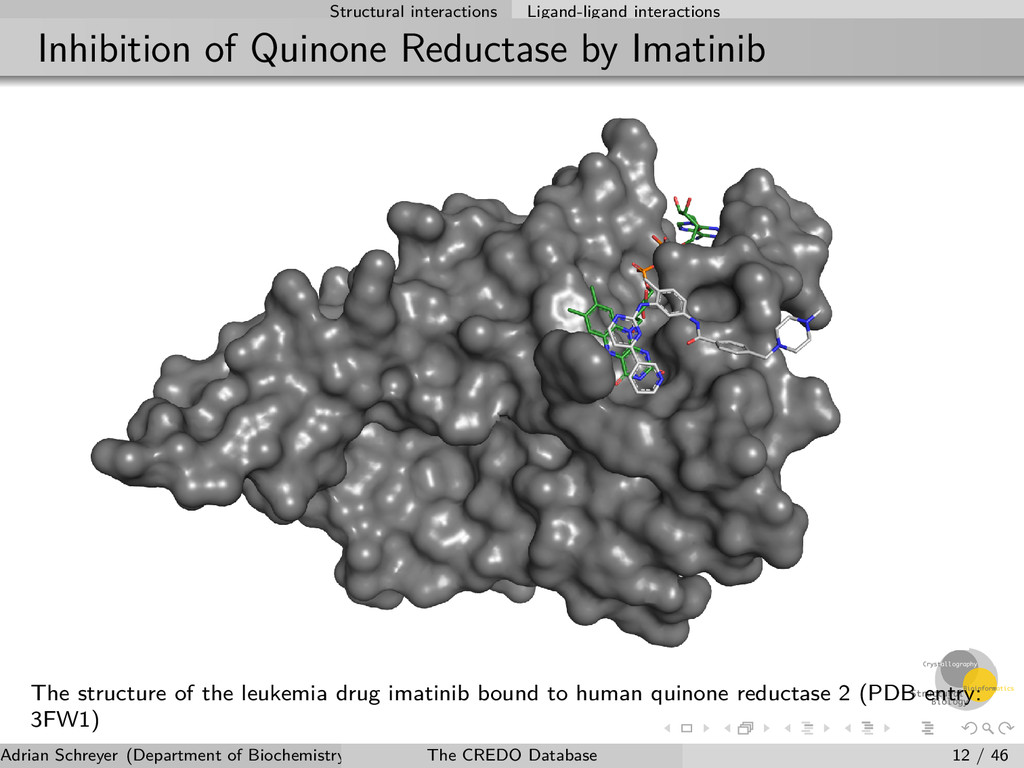
The structure of the leukemia drug imatinib bound to human quinone reductase 2 (PDB entry: 3FW1) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 12 / 46
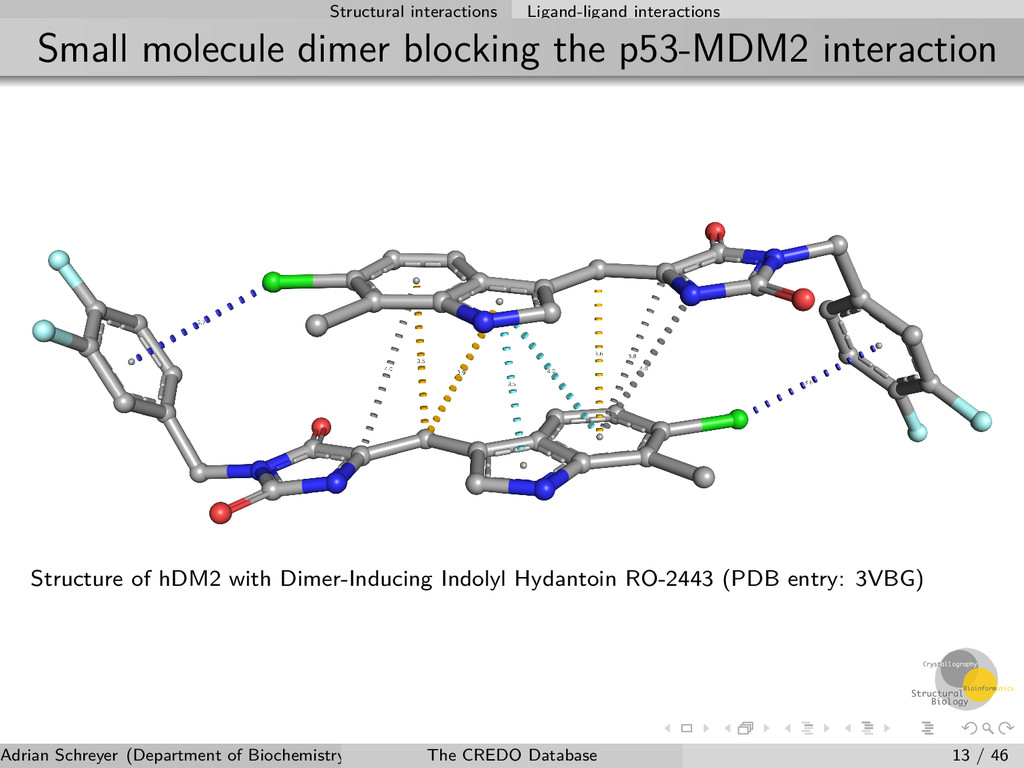
interaction Structure of hDM2 with Dimer-Inducing Indolyl Hydantoin RO-2443 (PDB entry: 3VBG) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 13 / 46
Fingerprints (SIFts) Aromatic ring interactions Ligand-ligand interactions Data Validation Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 14 / 46
All atomic data is retained (b-factors, occupancies) Boolean flags to identify missing/disordered/clashing residues and atoms Boolean flags to identify non-standard, modified and mutated amino acids Additional properties from mmCIF: resolution, r-factor, r-free, pH Ligand geometry (angles) can be problematic Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 15 / 46
index (DPI) Introduced by Cruickshank to estimate the uncertainty of atomic coordinates obtained by structural refinement of protein diffraction data Introduced to the virtual screening community by Goto Goto’s formula to calculate DPI σ(r, Bavg ) = 2.2N1/2 atoms V 1/2 a N−5/6 obs Rfree Goto’s formula to calculate theoretical DPI limit σ(r, Bavg ) = 0.22(1 + s)1/2V −1/2 m C−5/6Rfreed5/2 min Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 16 / 46
of missing regions and a secondary structure fragment (PDB entry: 2P33) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 17 / 46
Annotation of protein-ligand interactions SIFt clustering Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 18 / 46
pathways EC information is mapped onto protein chains KEGG data is used to identify metabolites and to link them to enzymes Ligands are labelled as substrate, product or cofactor (of the enzyme) Drug-target interactions Approved drugs are identified as well as all other compounds in the ChEMBL database Biological target information (UniProt) is taken from ChEMBL and DrugBank Drug-target interactions are identified Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 19 / 46
Potency of ligands Obtained from the latest version of the ChEMBL database Identified through a combination of document (PubMed), target (UniProt) and chemistry (UniChem) match Binding activities and ligand efficiencies (pKd, BEI, SEI) are linked to ligands where possible 6,848 unique activities for 6,505 unique ligands (28,943 pairs) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 20 / 46
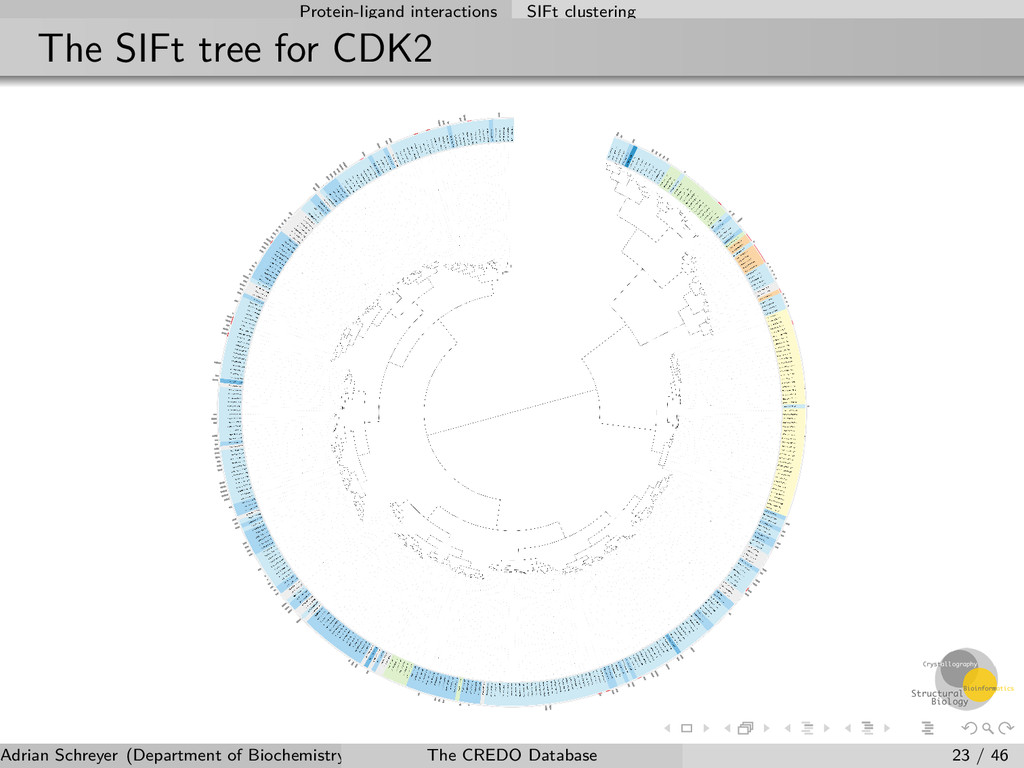
can be aligned to a given sequence system such as UniProt (or structural alignments) These alignments can be used for hierarchical clustering to compare interactions In CREDO this is done for all ligands that interact with proteins 2D and 3D similarities are calculated for terminal (leaf) nodes (always contain two ligands) Integrated into the website and API, phylogenetic trees can be visualised and browsed dynamically Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 22 / 46
and variations Sequence-to-structure mapping Structural variations affecting PDB residues and their interactions Binding site similarity searching Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 24 / 46
PDB chains Structure integration with function, taxonomy and sequence (SIFTS) initiative Maps UniProt sequences onto PDB residue sequences Provides further residue level annotation from the IntEnz, GO, Pfam, InterPro, SCOP, CATH and Pubmed databases Used to identify modified or mutated amino acids in protein chains Contains secondary structure information for each residue Transformed into relational format and linked to all residues in CREDO Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 25 / 46
domains onto protein chains Protein domain classifications from Pfam, CATH and SCOP are integrated into CREDO Mapped to protein chains, ligand binding sites, protein-protein interfaces etc. Pfam has the largest coverage by far 5,724 unique Pfam domains Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 26 / 46
secondary structure fragments The secondary structure information is used to create continuous fragments of secondary structure elements (SSE) in protein chains New fragment is identified after every change in secondary structure in the sequence of a polypeptide chain Tightly integrated with other CREDO entities Easily possible to get all SSEs interacting with a ligand or across a protein-protein interface Potential application in the context of peptidomimetic drugs and biologics Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 27 / 46
their interactions Outline 4 Protein sequences and variations Sequence-to-structure mapping Structural variations affecting PDB residues and their interactions Binding site similarity searching Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 28 / 46
their interactions Structural Variations in CREDO Identifying variations in protein structures Mapped onto residues in CREDO through sequence-to-structure mapping Can be easily queried and combined with other parameters Linked to EnsEMBL disease phenotypes 2,369 phenotypes can be linked to residues in CREDO Source databases included in EnsEMBL Variation dbSNP Catalogue Of Somatic Mutations In Cancer (COSMIC) Online Mendelian Inheritance in Man (OMIM) 1000 Genomes Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 29 / 46
their interactions Relevance: drug resistance in cancer C-KIT tyrosine kinase in complex with Imatinib (PDB entry: 1T46) with T670I Imatinib-resistant mutation. Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 30 / 46
Protein sequences and variations Sequence-to-structure mapping Structural variations affecting PDB residues and their interactions Binding site similarity searching Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 31 / 46
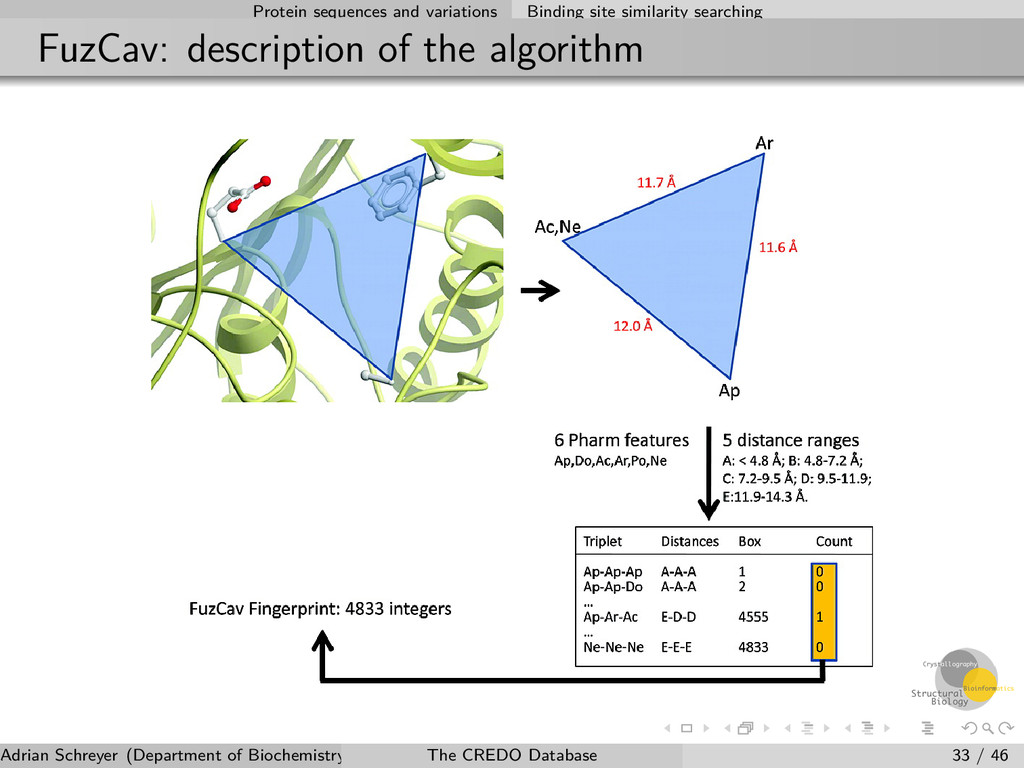
site similarity The FuzCav algorithm Alignment-free and very easy to calculate Based on pharmacophore triplet count to describe a ligand binding site Can detect local similarities between binding sites Performed natively on the server-side with PostgreSQL using numerical extension (pgeigen) Various similarity metrics can be used Calculated for all binding sites in CREDO Journal of Chemical Information and Modeling 2010 50 (1), 123-135 Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 32 / 46
Molecular descriptors RECAP fragmentation of chemical components Cheminformatics Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 34 / 46
Important to evaluate drug-likeness and filter molecules Feature counts, tPSA, XLogP, QED, ... Conformation-dependent Calculated for all bound ligands and their up to 200 modelled conformers Solvent-exluded and polar/apolar/total solvent-accessible surface areas Radius of gyration, Number of internal contacts Ultrafast-Shape Recognition (USR) moments as well as USRCAT Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 35 / 46
Chemistry and cheminformatics Molecular descriptors RECAP fragmentation of chemical components Cheminformatics Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 36 / 46
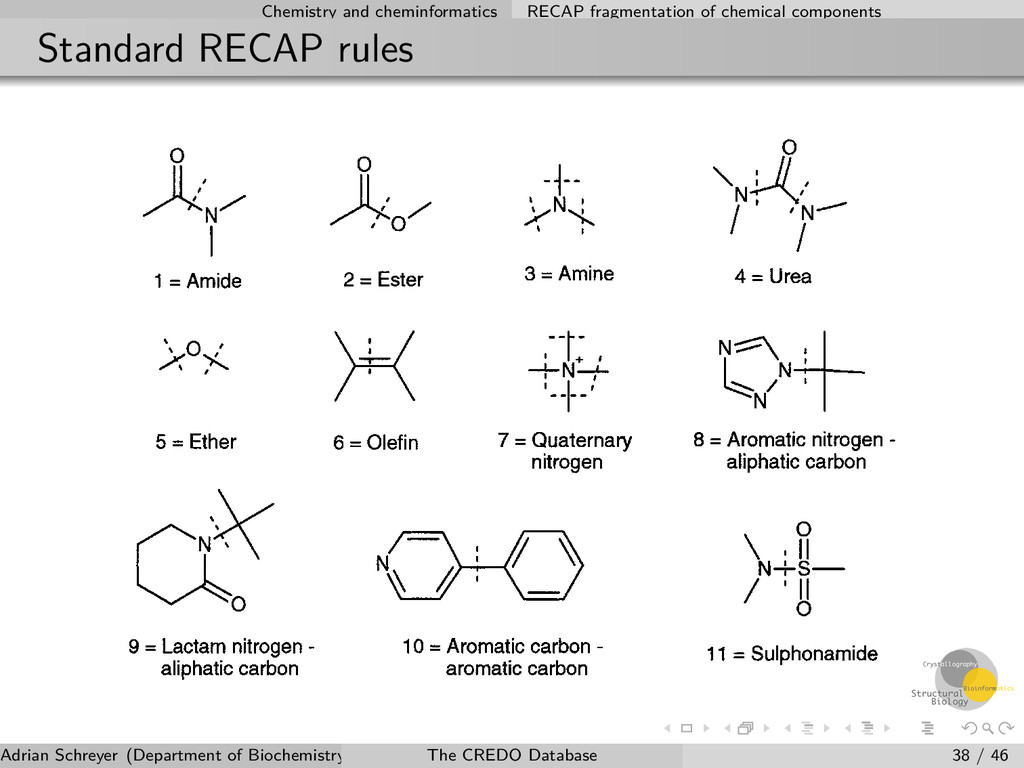
of chemical components Implementation of the algorithm The Retrosynthetic Combinatorial Analysis Procedure (RECAP) uses predefined bond types to cleave molecules into fragments A hierarchical and exhaustive fragmentation implementation is used in CREDO Hierarchy stored in the database and linked to chemical components New rules have been implemented to optimise fragmentation of natural products and endogenous compounds Existing rules have been extended (thioethers, thioesters,...) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 37 / 46
and ligands Analysing fragment interactions RECAP fragments are mapped back onto the ligands and their atoms of the original chemical components Therefore it is possible to analyse interactions on the fragment level Fragments can easily be filtered by their interactions, e.g. contact type or interactions with specific amino acids CREDO currently contains two measures to assess the contribution of a fragment to the interaction as a whole Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 39 / 46
Density (FCD) New measure to calculate fragment contributions Do all ligand fragments form an equal number of contacts or a single fragment dominate? Ratio between the number of contacts divided by the number of atoms for both the fragment and the whole ligand Number of contacts is simply the number of protein atoms within 4.5Å of the fragment Simple formula to calculate the Fragment Contact Density FCD = NFragment Contacts /NFragment Heavy atoms NLigand Contacts /NLigand Heavy atoms Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 40 / 46
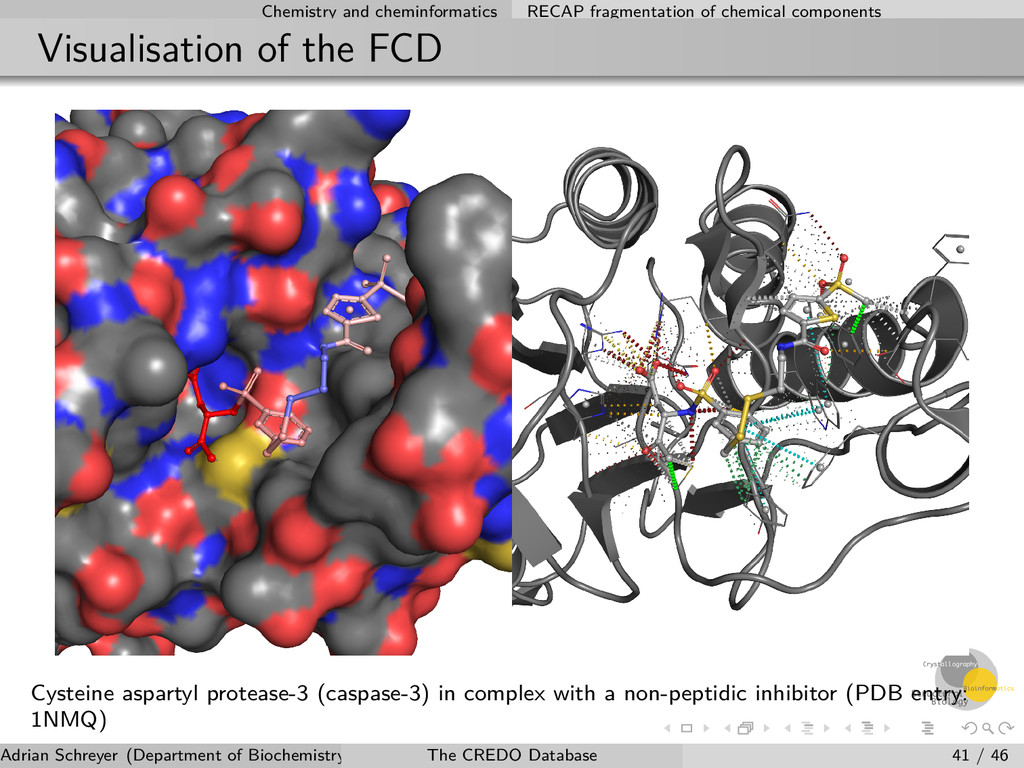
the FCD Cysteine aspartyl protease-3 (caspase-3) in complex with a non-peptidic inhibitor (PDB entry: 1NMQ) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 41 / 46
descriptors RECAP fragmentation of chemical components Cheminformatics Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 42 / 46
extension based on the OpenEye toolkits Implements commonly used cheminformatics routines Substructure, topological similarity, SMARTS, Murcko scaffolds, etc. Supports I/O of SMILES, SDF, OEB, IUPAC Fingerprint similarity metrics use SSE (POPCNT) Fingerprints can be indexed (GIST): 1.2M fingerprints, ordered result in less than 100 ms Very fast MCS search: 6500 structures < 100 ms (great with ChEMBL) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 43 / 46
USRCAT: an extension of USR USRCAT is an extension of Ultrafast Shape Recognition (USR) that includes pharmacophoric information into the moments Outperforms USR significantly in a virtual screening benchmark (using DUD-E) Implemented natively into the database: can be used in any SQL query (limit to specific family | include chemical graph similarity) Average screening performance of 5.3M conformers (moments) per second (including sorting) Currently used with all PDB chemical components and ZINC drug-like set (12M compounds, 200M+ conformers) Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 44 / 46
Can be used to browse and search data in CREDO Biological assemblies can be visualised directly, including visualisation of contacts and highlighting of mutations (WebGL) Downloads of selected data sets, e.g. kinases RESTful Web service Most resources of the service can be queried programmaticly through GET or POST requests Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 45 / 46
information and updates Web interface: http://www-cryst.bioc.cam.ac.uk/credo Blog: http://blog.adrianschreyer.com Twitter: http://twitter.com/credodb Adrian Schreyer (Department of Biochemistry, University of Cambridge) The CREDO Database 46 / 46
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}