Templates for Predicting Protein – DNA Interactions Jamie Duke1,2 and Carlos Camacho3 1Bioengineering and Bioinformatics Summer Institute, Department of Computational Biology, University of Pittsburgh, Pittsburgh, PA 15261 2Department of Biological Sciences, Rochester Institute of Technology, Rochester, NY 14623 3Department of Computational Biology, University of Pittsburgh, Pittsburgh, PA 15261 Goals Investigate the diversity of the EGR family of proteins Carry out homology modeling between resolved structures and known human EGR proteins Test the structures with protein – DNA docking algorithms to determine the specific protein – DNA interactions
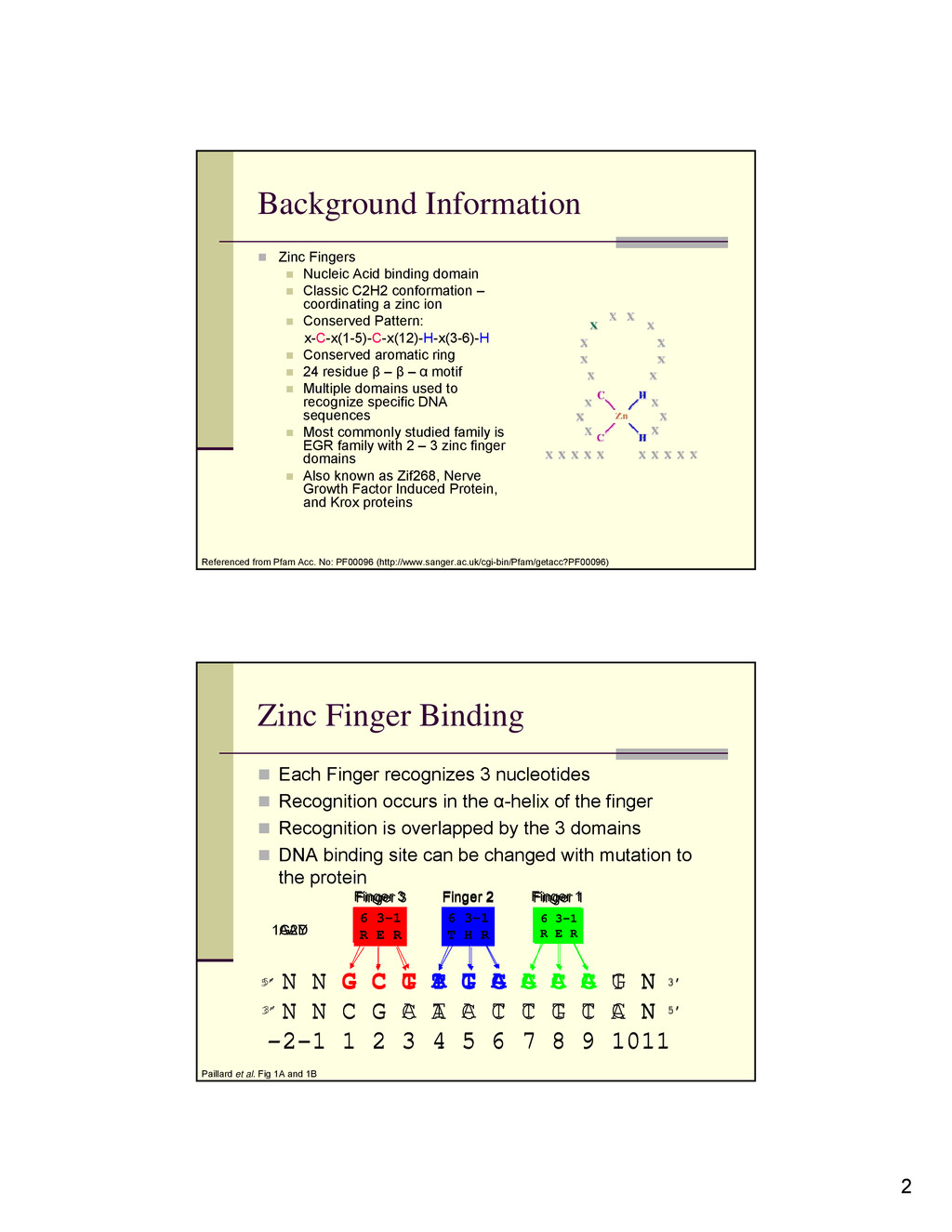
domain Classic C2H2 conformation – coordinating a zinc ion Conserved Pattern: x-C-x(1-5)-C-x(12)-H-x(3-6)-H Conserved aromatic ring 24 residue β – β – α motif Multiple domains used to recognize specific DNA sequences Most commonly studied family is EGR family with 2 – 3 zinc finger domains Also known as Zif268, Nerve Growth Factor Induced Protein, and Krox proteins Referenced from Pfam Acc. No: PF00096 (http://www.sanger.ac.uk/cgi-bin/Pfam/getacc?PF00096) 5’ 3’ 5’ 3’ 6 3-1 R T T 6 3-1 Q G Q 6 3-1 T N Q N N G C T A T A A A A G N N N C G A T A T T T T C N -2-1 1 2 3 4 5 6 7 8 9 1011 Finger 3 Finger 2 Finger 1 Zinc Finger Binding Each Finger recognizes 3 nucleotides Recognition occurs in the α-helix of the finger Recognition is overlapped by the 3 domains DNA binding site can be changed with mutation to the protein Paillard et al. Fig 1A and 1B 5’ 3’ 6 3-1 R E R 6 3-1 T H R 6 3-1 R E R N N G C G T G G G C G T N N N C G C A C C C G C A N -2-1 1 2 3 4 5 6 7 8 9 1011 Finger 3 Finger 2 Finger 1 5’ 3’ 1AAY 1G2D
zf-C2H2 proteins in the Protein Data Bank 11 structures were applicable to our interests Of the 42 structures: 20 were from x-ray crystallography 22 were developed through NMR At least 15 were duplicate structures We only considered structures that were developed through x-ray crystallography and had either 2 or 3 zinc fingers, as they would belong to the EGR family Homology Modeling We chose two proteins with known structures to perform homology modeling, 1G2D and 1AAY Allows us to compare the predicted structure against the known structure to determine the accuracy of the prediction A Zif268 variant (1G2D) was selected for the target of the homology modeling, with the template being Zif268 (1AAY) The 1G2D recognizes the DNA Sequence: 5’– GCTATAAAA – 3’ The sequences are 83% similar, with 81% sequence identity 1AAY MERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFS MERPYACPVESCDRRFS+ L HIRIHTGQKPFQCRICMRNFS 1G2D MERPYACPVESCDRRFSQKTNLDTHIRIHTGQKPFQCRICMRNFS 1AAY RSDHLTTHIRTHTGEKPFACDICGRKFARSDERKRHTKIHLRQKD + L HIRTHTGEKPFACDICGRKFA R RHTKIHLRQKD 1G2D QHTGLNQHIRTHTGEKPFACDICGRKFATLHTRDRHTKIHLRQKD
3 and 6 in the α-helix The Consensus Server, developed in part by Dr. Camacho, was used to perform the homology modeling (http://structure.bu.edu/cgi- bin/consensus/consensus.cgi) Since threading algorithms are used in the Consensus method, the side chains of amino acids can only be predicted to the extent of the corresponding amino acid from the template. Serine Æ Lysine the method can only place Cα and Cβ atoms, leaving four carbon atoms positions undeterminable. CHARMM was used to complete the side chains Side Chain Relaxation via Molecular Dynamic Simulations We chose to relax the side chains for each domain independently to find the most favorable state Simulations were run using a constrained backbone to conserve the structure that was predicted in the previous step Run-time totaled of 4.2 ns for each domain 200 ps for system equilibration, Each time step was 2 fs This simulation did not take into account ions and without the DNA present We were particularly interested in the states of the three residues involved in DNA recognition
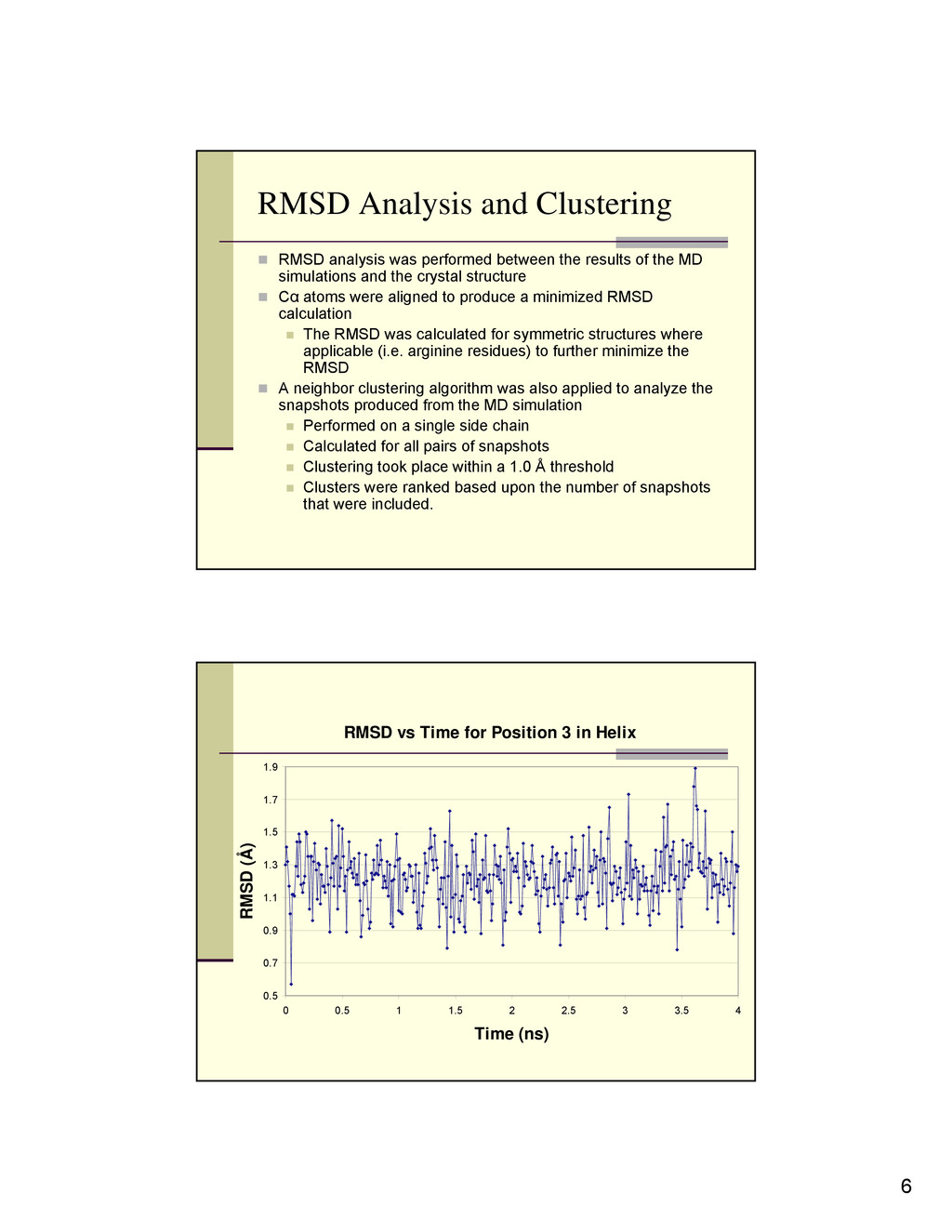
between the results of the MD simulations and the crystal structure Cα atoms were aligned to produce a minimized RMSD calculation The RMSD was calculated for symmetric structures where applicable (i.e. arginine residues) to further minimize the RMSD A neighbor clustering algorithm was also applied to analyze the snapshots produced from the MD simulation Performed on a single side chain Calculated for all pairs of snapshots Clustering took place within a 1.0 Å threshold Clusters were ranked based upon the number of snapshots that were included. RMSD vs Time for Position 3 in Helix 0.5 0.7 0.9 1.1 1.3 1.5 1.7 1.9 0 0.5 1 1.5 2 2.5 3 3.5 4 Time (ns) RMSD (Å)
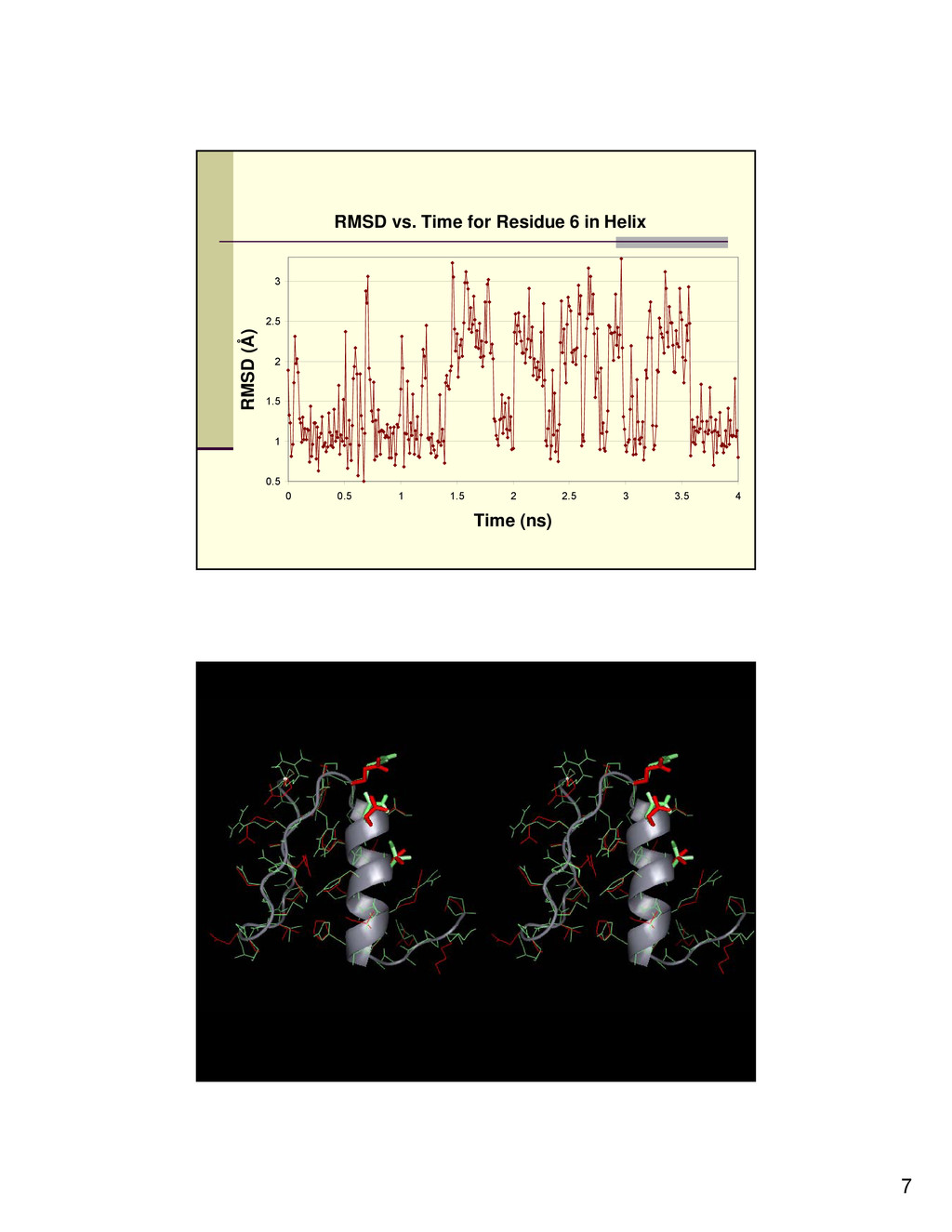
determined that most of the residues reach an equilibrium point that is highly similar to the crystal structure. Cluster analysis revealed that the cluster with the most amount of neighbors is generally highly similar to the crystal structure. There are a few residues that are seen in the simulation that seem to fluctuate between two states, as can be seen in Figure 3. We believe that this fluctuation may be correlated to the mechanism by which the protein recognizes the DNA.
two other situations: Modeling Zif268 (1AAY) using the Zif268 variant (1G2D) as a template Modeling Designed Zinc Finger (1MEY) using Zif268 as the template Shares 49% identity with 1AAY Shares 47% identity with 1G2D Preliminary results and analysis show similar findings to 1G2D modeled after 1AAY Conclusions and Future Applications Through this method we are able to effectively determine a homology model of zinc finger proteins, more specifically zinc finger proteins in the EGR family. The modeled side chains are found to be in a state that is similar to the crystal structure, even when in an unbound state, which is particularly important for the key residues involved in DNA recognition. Since the modeled domains are in a desirable conformation, it is possible to perform docking experiments with homology modeled zinc fingers, which is currently being done using an DNA-protein docking algorithm developed in the lab. Future applications include modeling EGR proteins with an undetermined structure to see if the model is able to recognize the proper DNA sequence.
Champ BBSI – Department of Computational Biology, University of Pittsburgh NIH – NSF References J.C. Prasad, S.R. Comeau, S. Vajda, and C.J. Camacho. Consensus alignment for reliable framework prediction in homology modeling. Bioinformatics 2003 19: 1682-1691. Paillard G., Deremble C., Lavery R. Looking into DNA Recognition: Zinc Finger Binding Specificity. Nucleic Acids Research 2004 32: 6673-6682. A. Bateman, L. Coin, R. Durbin, R.D. Finn, V. Hollich, S. Griffiths-Jones, A. Khanna, M. Marshall, S. Moxon, E.L.L. Sonnhammer, D.J. Studholme, C. Yeats, S.R. Eddy. The Pfam Protein Families Database. Nucleic Acids Research: Database Issue 2004 32: D138- D141.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}