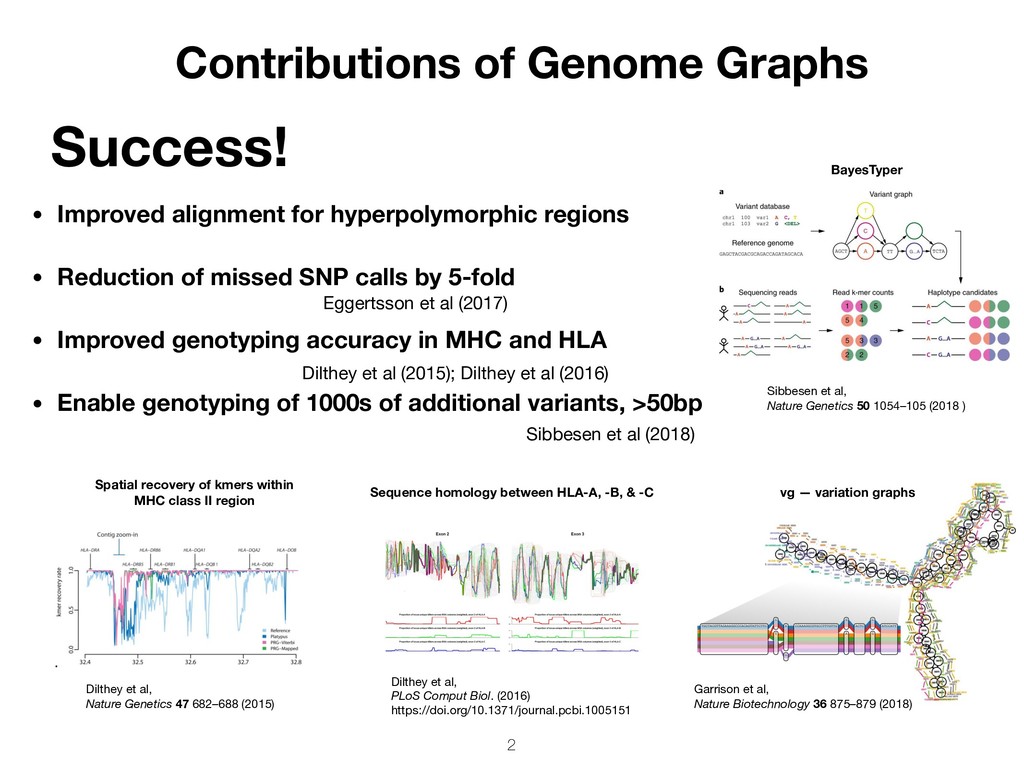
SNP calls by 5-fold • Improved genotyping accuracy in MHC and HLA • Enable genotyping of 1000s of additional variants, >50bp Success! Contributions of Genome Graphs Garrison et al, Nature Biotechnology 36 875–879 (2018) Sequence homology between HLA-A, -B, & -C Dilthey et al, PLoS Comput Biol. (2016) https://doi.org/10.1371/journal.pcbi.1005151 2 vg — variation graphs BayesTyper Sibbesen et al, Nature Genetics 50 1054–105 (2018 ) Dilthey et al, Nature Genetics 47 682–688 (2015) Spatial recovery of kmers within MHC class II region Sibbesen et al (2018) Dilthey et al (2015); Dilthey et al (2016) Eggertsson et al (2017)
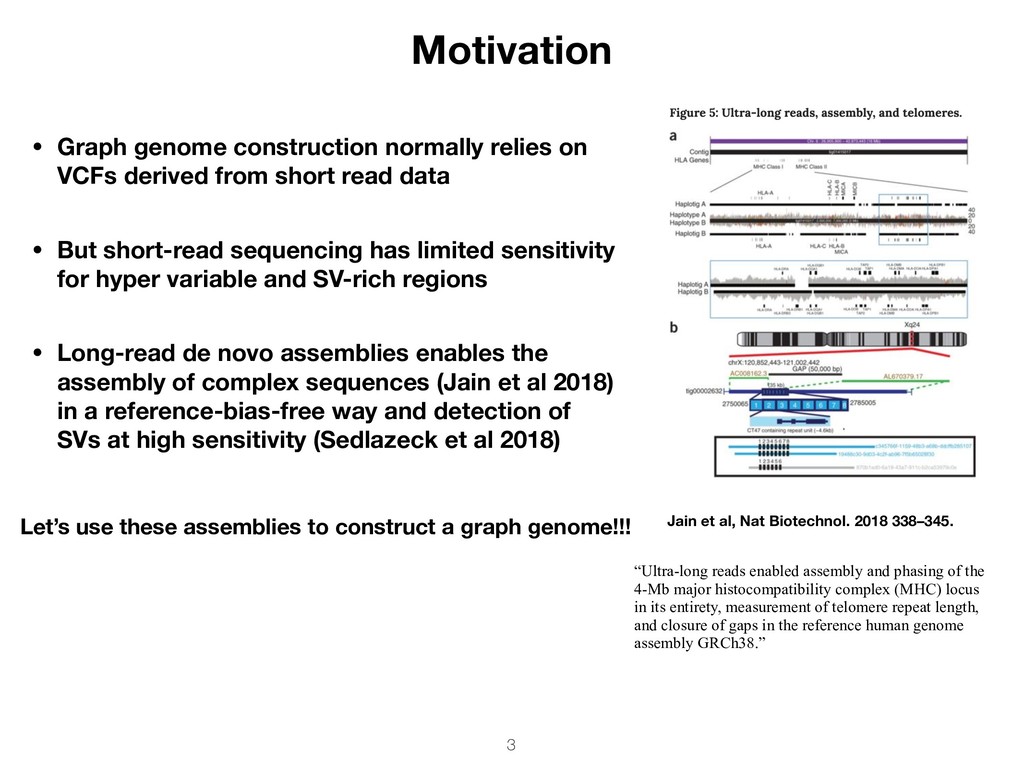
4-Mb major histocompatibility complex (MHC) locus in its entirety, measurement of telomere repeat length, and closure of gaps in the reference human genome assembly GRCh38.” • Graph genome construction normally relies on VCFs derived from short read data • But short-read sequencing has limited sensitivity for hyper variable and SV-rich regions • Long-read de novo assemblies enables the assembly of complex sequences (Jain et al 2018) in a reference-bias-free way and detection of SVs at high sensitivity (Sedlazeck et al 2018) Jain et al, Nat Biotechnol. 2018 338–345. Let’s use these assemblies to construct a graph genome!!!
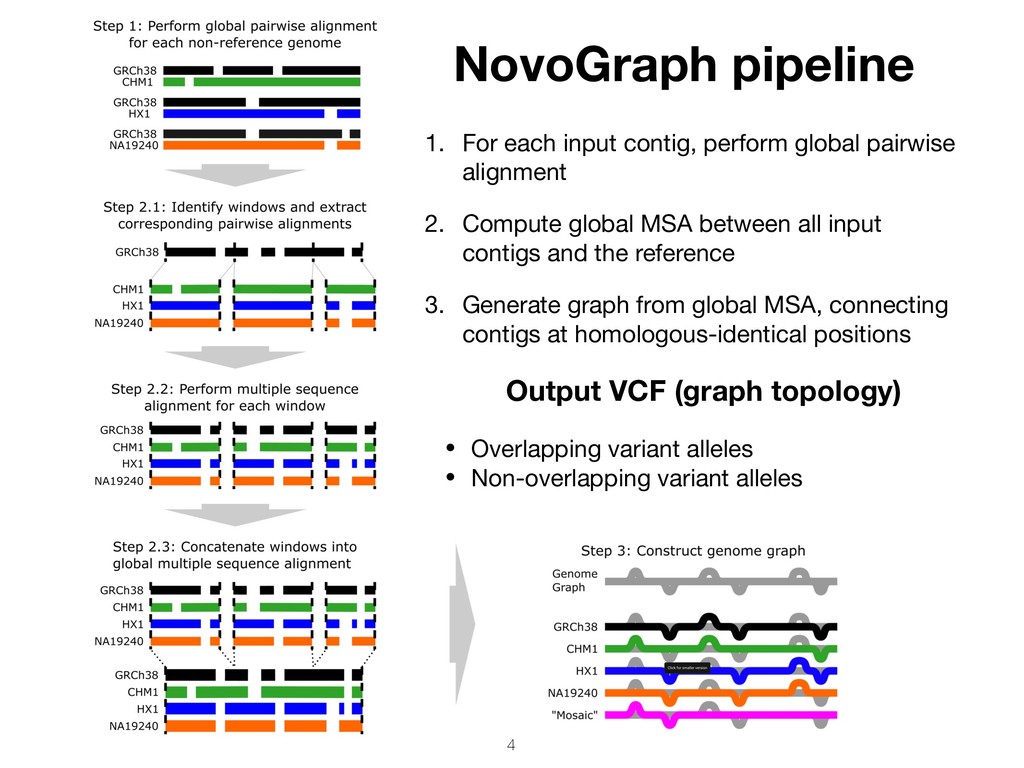
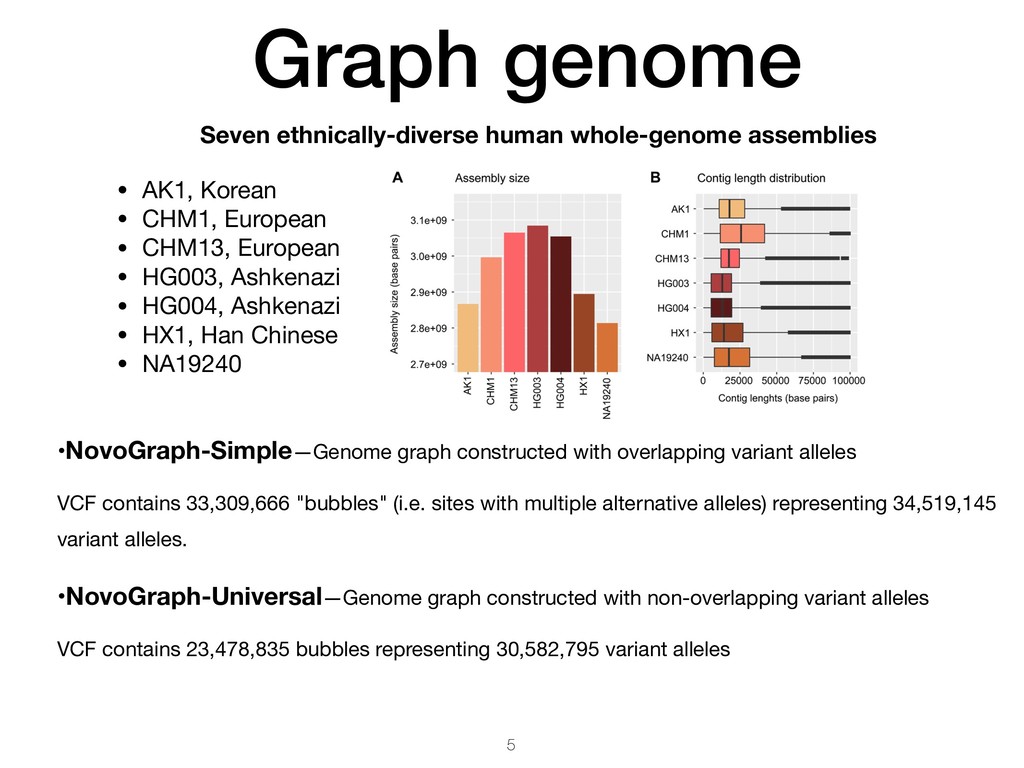
pairwise alignment 2. Compute global MSA between all input contigs and the reference 3. Generate graph from global MSA, connecting contigs at homologous-identical positions Output VCF (graph topology) • Overlapping variant alleles • Non-overlapping variant alleles
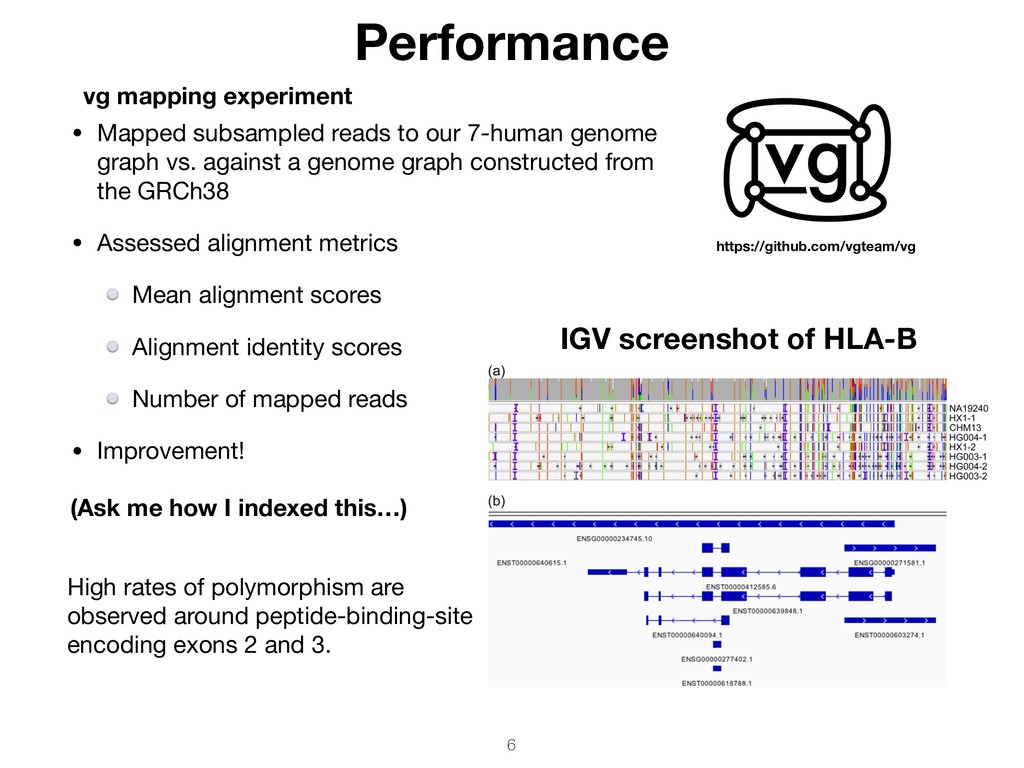
vs. against a genome graph constructed from the GRCh38 • Assessed alignment metrics Mean alignment scores Alignment identity scores Number of mapped reads • Improvement! Performance IGV screenshot of HLA-B High rates of polymorphism are observed around peptide-binding-site encoding exons 2 and 3. vg mapping experiment (Ask me how I indexed this…) https://github.com/vgteam/vg
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}