#BioIT19 NovoGraph Genome graph construction from multiple long-read de novo assemblies A genome graph representation of seven ethnically diverse whole human genomes 1
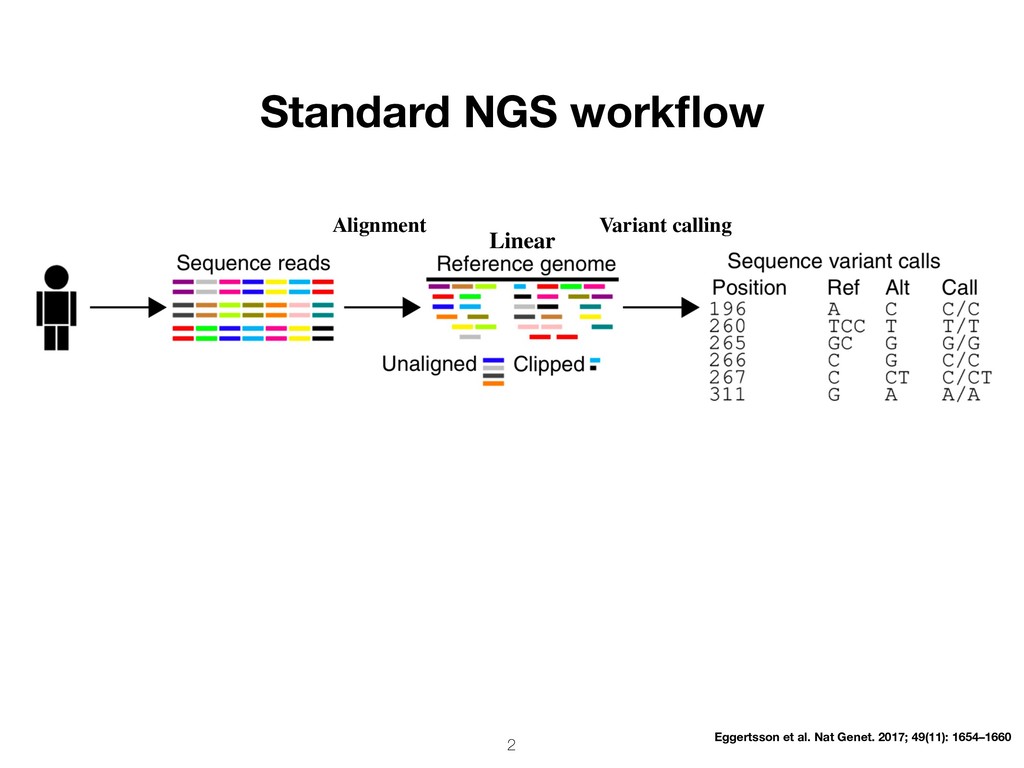
approach fails for reads from hyperpolymorphic regions (e.g. MHC) or regions with large and/or complex SVs • Estimated >1% of human genome inaccessible with classic approach. Dilthey et al, Nat Genet. 2015; 47(6): 682–688 • The linear reference (GRCh 38) is a single consensus haploid genome. This biases alignment towards reference allele, results in inaccurate alignments, and skews genotyping accuracy. • Alignment not aware of sequence variation between/within subpopulations • African pan-genome of 910 individuals contains ~10% more DNA than the current human reference genome. Sherman et al. Nat Genet. 2019; 30–35 (2019)
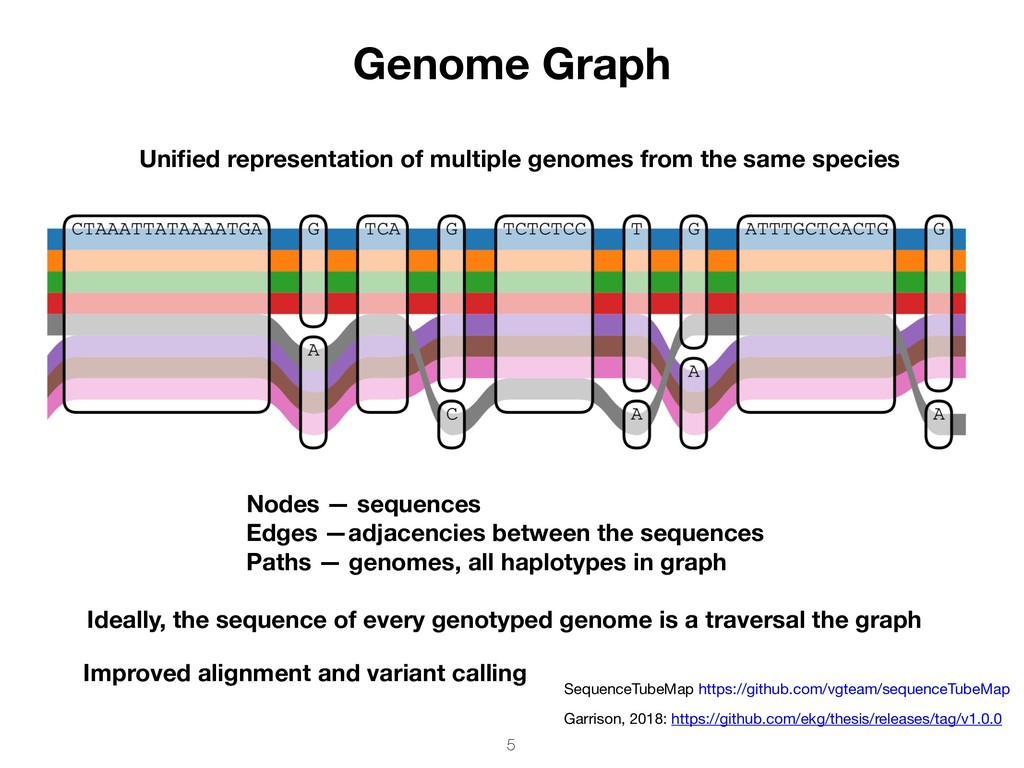
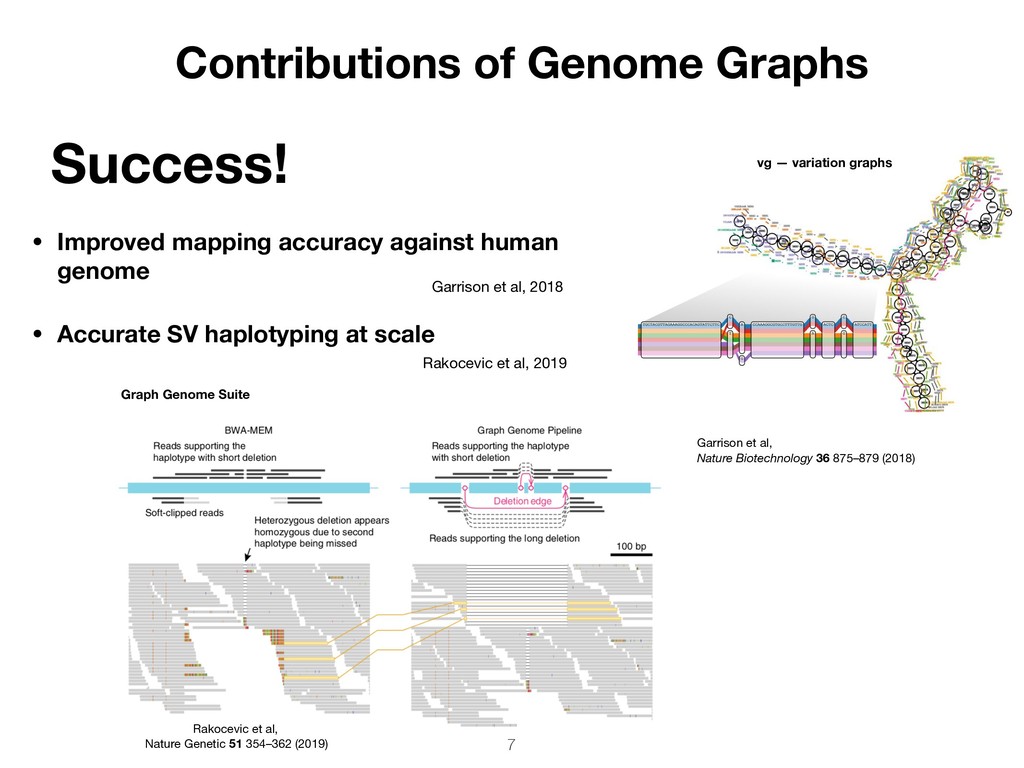
sequences Paths — genomes, all haplotypes in graph Ideally, the sequence of every genotyped genome is a traversal the graph SequenceTubeMap https://github.com/vgteam/sequenceTubeMap Garrison, 2018: https://github.com/ekg/thesis/releases/tag/v1.0.0 Unified representation of multiple genomes from the same species Improved alignment and variant calling
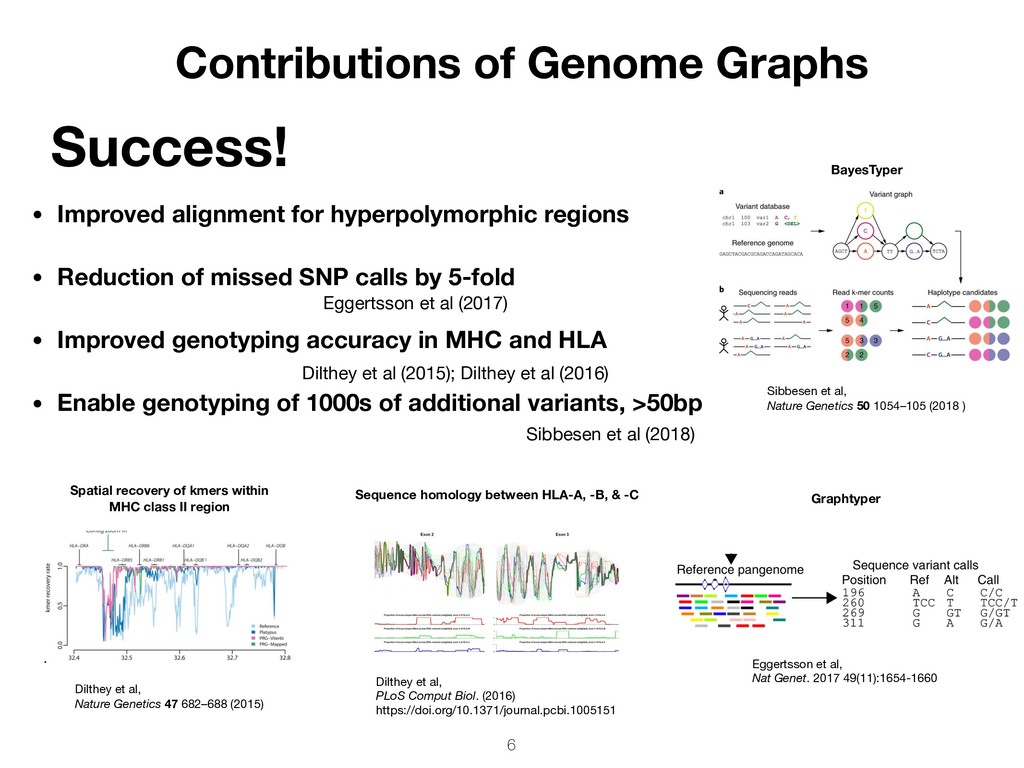
SNP calls by 5-fold • Improved genotyping accuracy in MHC and HLA • Enable genotyping of 1000s of additional variants, >50bp Success! Contributions of Genome Graphs Eggertsson et al, Nat Genet. 2017 49(11):1654-1660 Sequence homology between HLA-A, -B, & -C Dilthey et al, PLoS Comput Biol. (2016) https://doi.org/10.1371/journal.pcbi.1005151 6 Graphtyper BayesTyper Sibbesen et al, Nature Genetics 50 1054–105 (2018 ) Dilthey et al, Nature Genetics 47 682–688 (2015) Spatial recovery of kmers within MHC class II region Sibbesen et al (2018) Dilthey et al (2015); Dilthey et al (2016) Eggertsson et al (2017)
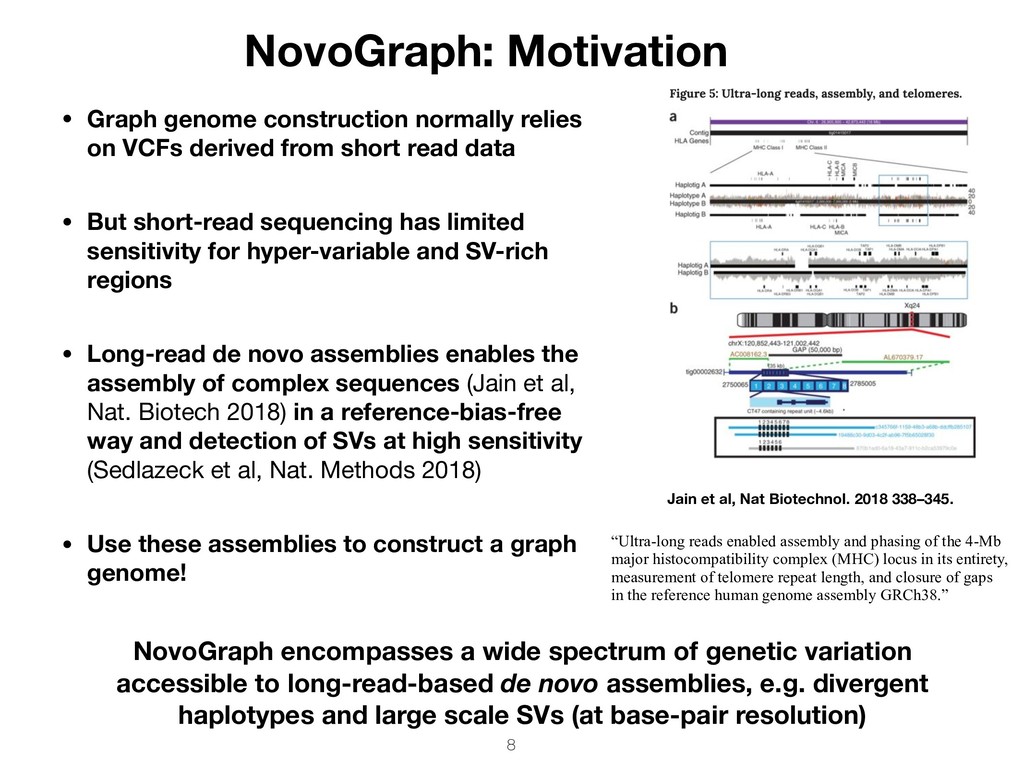
the 4-Mb major histocompatibility complex (MHC) locus in its entirety, measurement of telomere repeat length, and closure of gaps in the reference human genome assembly GRCh38.” • Graph genome construction normally relies on VCFs derived from short read data • But short-read sequencing has limited sensitivity for hyper-variable and SV-rich regions • Long-read de novo assemblies enables the assembly of complex sequences (Jain et al, Nat. Biotech 2018) in a reference-bias-free way and detection of SVs at high sensitivity (Sedlazeck et al, Nat. Methods 2018) • Use these assemblies to construct a graph genome! Jain et al, Nat Biotechnol. 2018 338–345. NovoGraph encompasses a wide spectrum of genetic variation accessible to long-read-based de novo assemblies, e.g. divergent haplotypes and large scale SVs (at base-pair resolution)
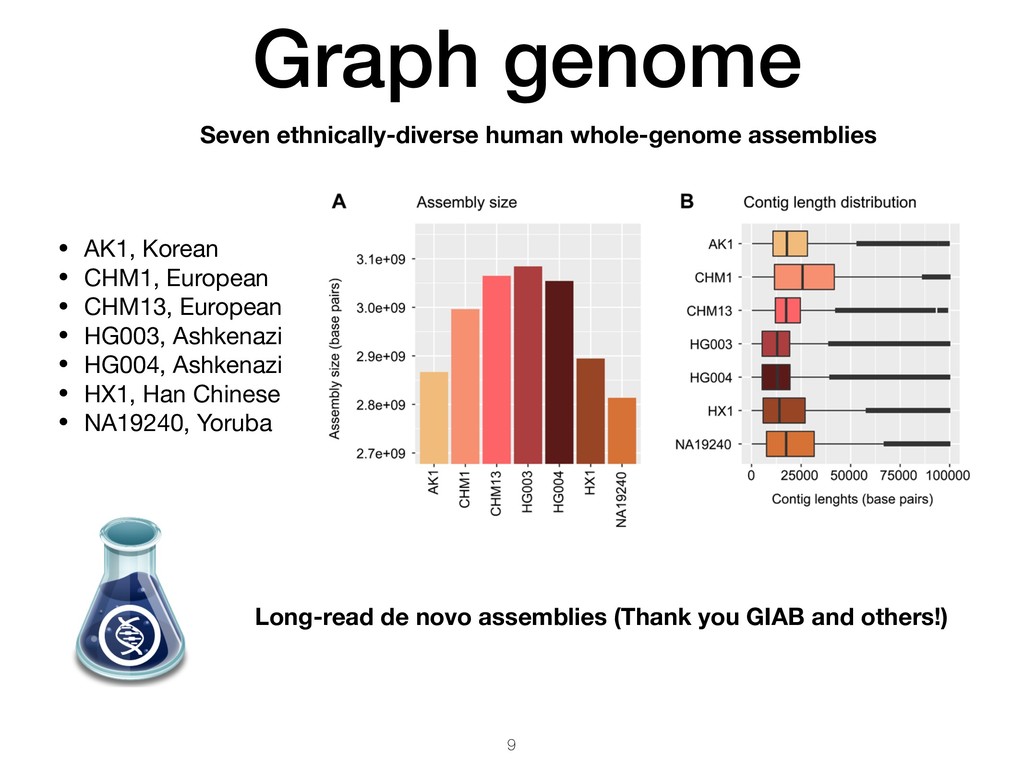
Korean • CHM1, European • CHM13, European • HG003, Ashkenazi • HG004, Ashkenazi • HX1, Han Chinese • NA19240, Yoruba Long-read de novo assemblies (Thank you GIAB and others!)
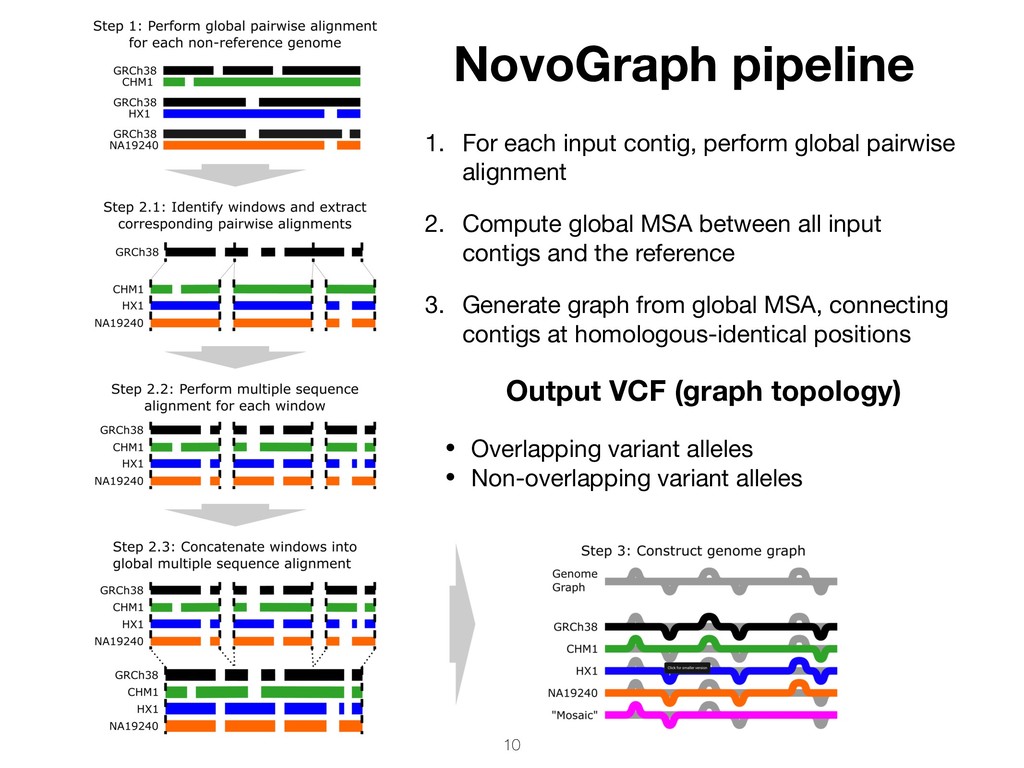
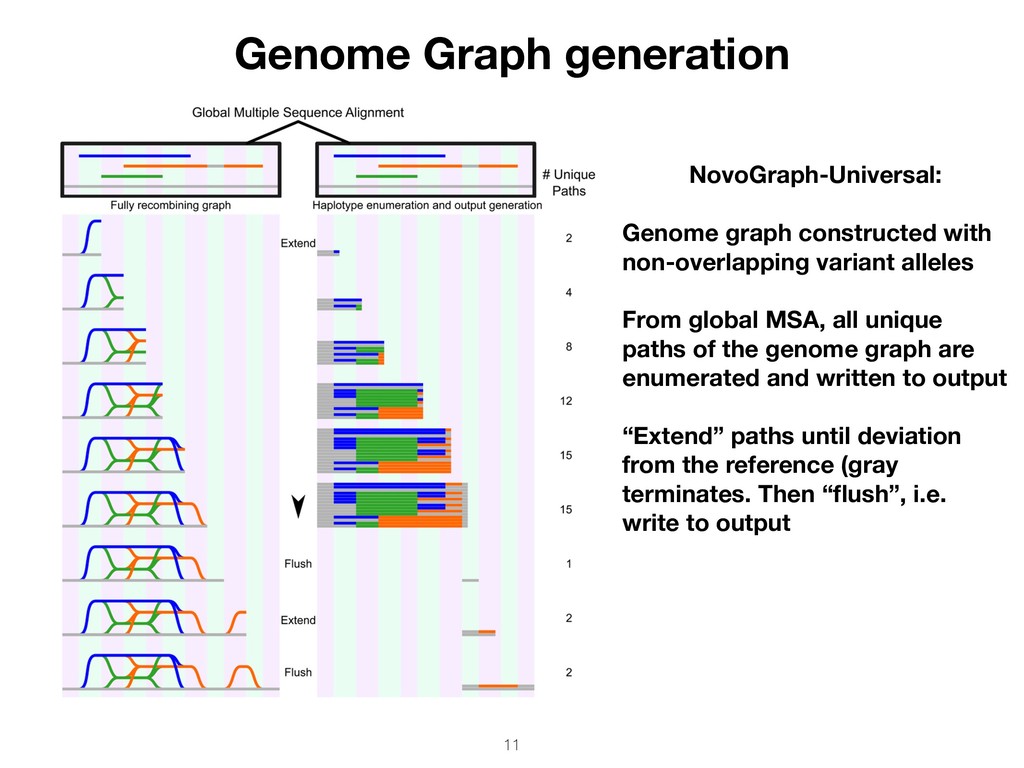
pairwise alignment 2. Compute global MSA between all input contigs and the reference 3. Generate graph from global MSA, connecting contigs at homologous-identical positions Output VCF (graph topology) • Overlapping variant alleles • Non-overlapping variant alleles
variant alleles From global MSA, all unique paths of the genome graph are enumerated and written to output “Extend” paths until deviation from the reference (gray terminates. Then “flush”, i.e. write to output
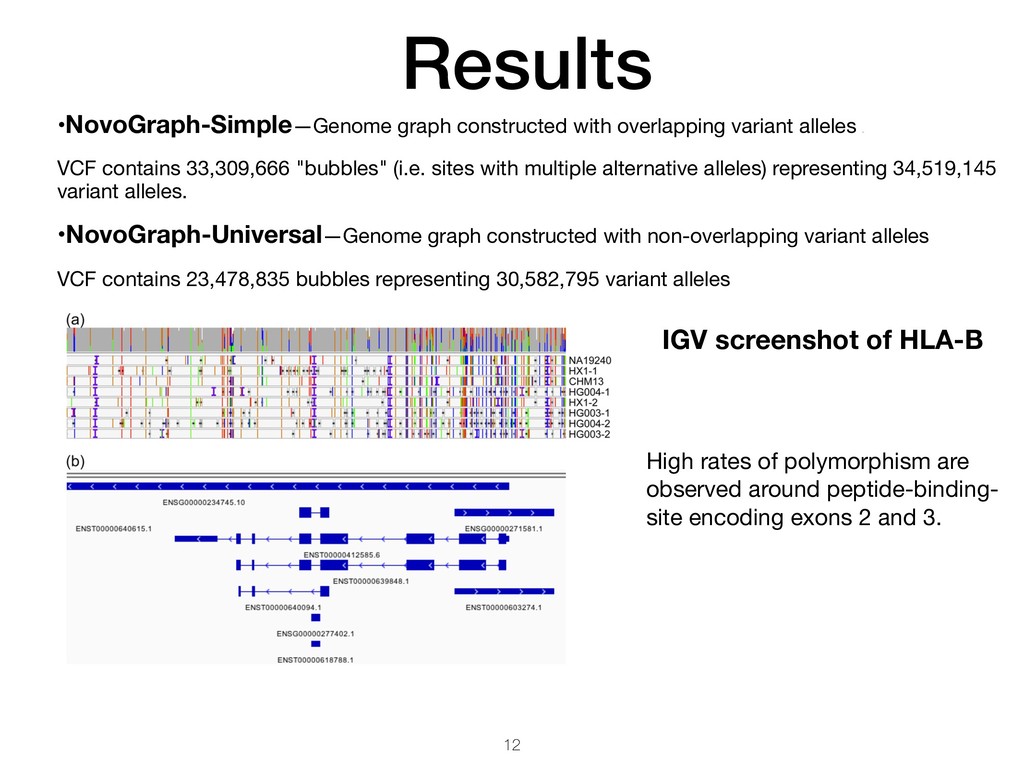
VCF contains 33,309,666 "bubbles" (i.e. sites with multiple alternative alleles) representing 34,519,145 variant alleles. •NovoGraph-Universal—Genome graph constructed with non-overlapping variant alleles VCF contains 23,478,835 bubbles representing 30,582,795 variant alleles IGV screenshot of HLA-B High rates of polymorphism are observed around peptide-binding- site encoding exons 2 and 3.
vs. against a genome graph constructed from the GRCh38 • Assessed alignment metrics Mean alignment scores Alignment identity scores Number of mapped reads • Improvement! Performance vg mapping experiment https://github.com/vgteam/vg As expected, mapping against the genome graph increases mean alignment scores and alignment identities, albeit at a small reduction in the number of mapped reads. Genome Graph Reference Genome Mean Scores 108.859 108.100 Mean Identity Value 0.9913 0.9891 Total Mapped Reads 31125004 31138410
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}