Center for Functional Cancer Epigenetics Department of Medical Oncology Department of Biostatistics and Computational Biology Dana-Farber Cancer Institute May 29, 2015
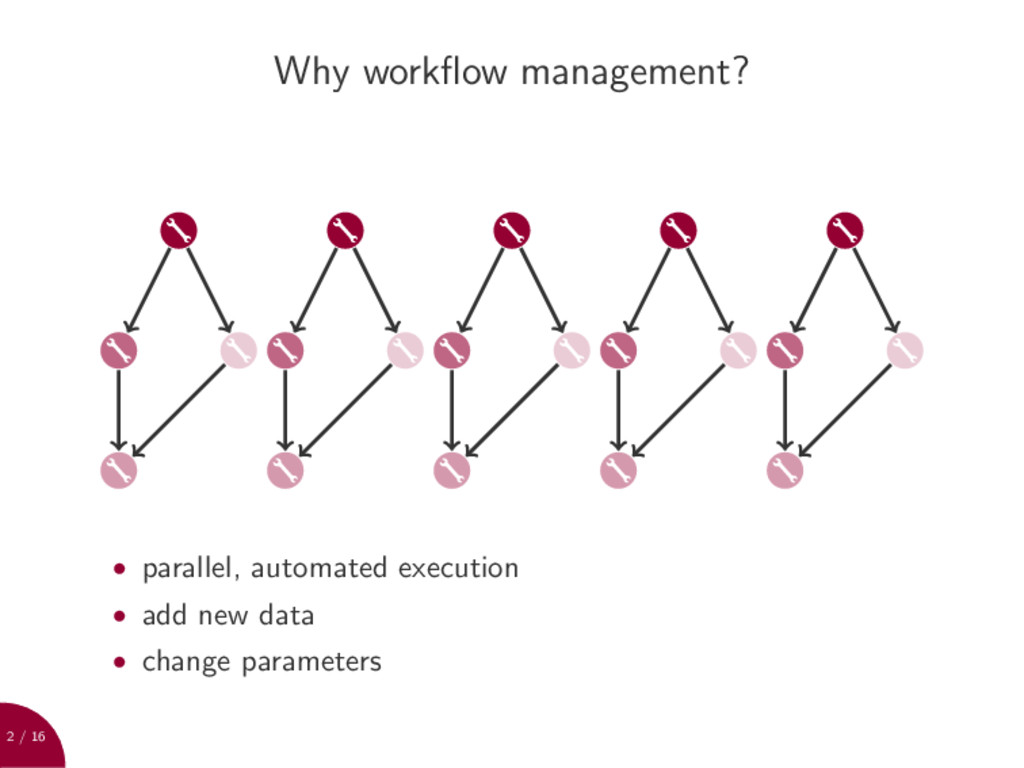
Make: • text based • rule paradigm • lightweight And what not: • cryptic syntax • limited scripting • missing support for multiple output files and wildcards • no cluster support
input : a=” path / to /{ sample }. t x t ” output : b=” path / to /{ sample }. column1 . t x t ” s h e l l : ” cut - f1 < { input . a} > { output . b}”
input : a=” path / to /{ sample }. t x t ” output : b=” path / to /{ sample }. column1 . t x t ” run : with open ( output . b , ”w” ) as out : f o r l in csv . reader ( open ( input . a ) ) : p r i n t ( l [ 0 ] , f i l e=out )
bam for each sample r u l e a l l : i np u t : ” 500.bam” , ” 501.bam” , ” 502.bam” , ” 503.bam” # map reads r u l e map : i np u t : r e f=” r e f e r e n c e . f a s t a ” , index=” r e f e r e n c e . bwt” , reads=”{sample }. f a s t q ” output : ”{sample }.bam” t h r e a d s : 8 s h e l l : ”bwa mem - t { t h r e a d s } { i n pu t . r e f } { i n pu t . reads} | ” # refer to threads and input ” samtools view - Sbh - > {output}” # refer to output files # create an index r u l e index : i np u t : ” r e f e r e n c e . f a s t a ” output : ” r e f e r e n c e . bwt” s h e l l : ”bwa index { i n p u t }”
501 502 503” . s p l i t () # require a bam for each sample r u l e a l l : i np u t : expand ( ”{sample }.bam” , sample=SAMPLES) # map reads r u l e map : i np u t : r e f=” r e f e r e n c e . f a s t a ” , index=” r e f e r e n c e . bwt” , reads=”{sample }. f a s t q ” output : ”{sample }.bam” t h r e a d s : 8 s h e l l : ”bwa mem - t { t h r e a d s } { i n pu t . r e f } { i np u t . reads} | ” # refer to threads and input ” samtools view - Sbh - > {output}” # refer to output files # create an index r u l e index : i np u t : ” r e f e r e n c e . f a s t a ” output : ” r e f e r e n c e . bwt” s h e l l : ”bwa index { i n p u t }”
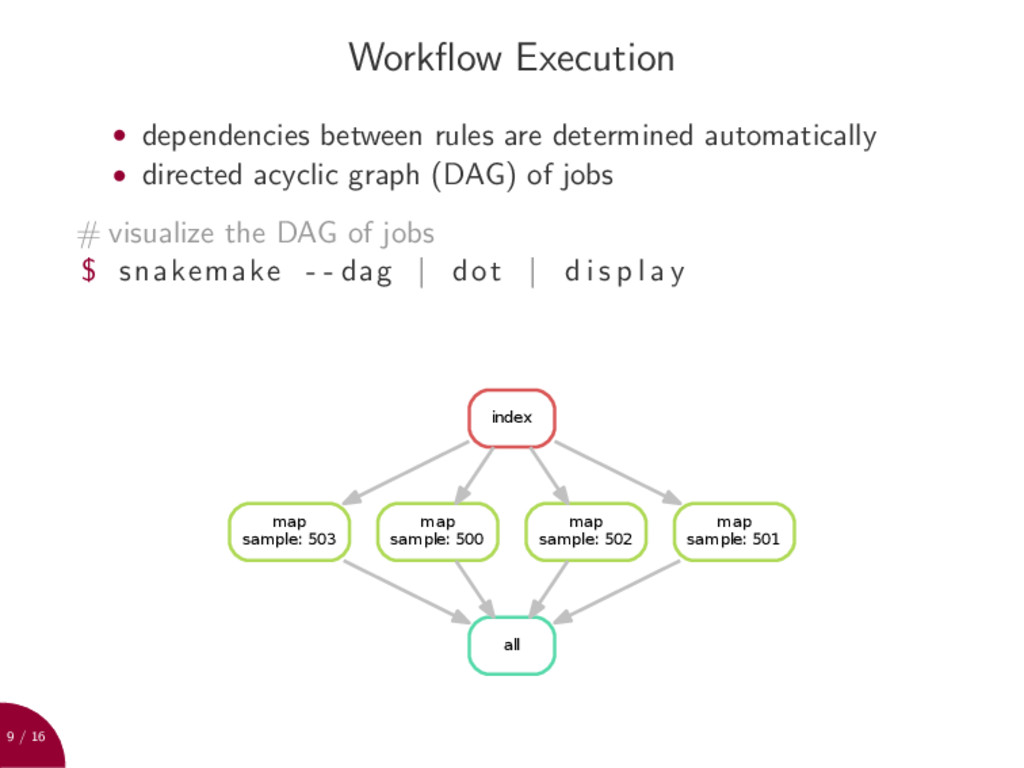
determined automatically • directed acyclic graph (DAG) of jobs # visualize the DAG of jobs $ snakemake - - dag | dot | d i s p l a y map sample: 503 all map sample: 500 map sample: 502 map sample: 501 index
DAG can be parallelized • only outdated or missing files are created # perform a dry-run $ snakemake - n # execute the workflow using 8 cores $ snakemake - - c o r e s 8 # execute the workflow on a cluster (with up to 20 jobs) $ snakemake - - j o b s 20 - - c l u s t e r ” qsub - pe threaded { t h r e a d s }” # execute the workflow on a cluster using the DRMAA API $ snakemake - - j o b s 20 - - drmaa
g f i l e : ” c o n f i g . yaml” r u l e map : i np u t : r e f=” r e f e r e n c e . f a s t a ” , index=” r e f e r e n c e . bwt” , reads=”{sample }. f a s t q ” output : ”{sample }.bam” log : ” lo g /{sample }. log ” # specify log files v e r s i o n : s h e l l ( ”bwa 2>&1 | grep Version ” ) # record used version r e s o u r c e s : mem=5 # add custom resources t h r e a d s : 8 s h e l l : ”bwa mem - k { c o n f i g [ s e e d l e n ]} - t { t h r e a d s } { i n pu t . r e f } { i np u t . reads} | ” ” samtools view - Sbh - > {output} 2> {log}”
- - r e s o u r c e s mem=5 - - cores 16 # prioritize a target $ snakemake - - p r i o r i t i z e 500.bam Maximize the number of running jobs with respect to • priority • number of descendants • input size while not exceeding • provided cores • provided resources A multi-dimensional knapsack problem.
. s p l i t () # define subworkflow subworkflow mapping : workdir : ” . . / mapping” r u l e a l l : i np u t : expand ( ”{sample}/ r e s u l t s . xprs ” , sample=SAMPLES) # estimate transcript expressions r u l e e x p r e s s : i np u t : REF , mapping ( ”{sample }.bam” ) # refer to output of subworkflow output : ”{sample}/ r e s u l t s . xprs ” s h e l l : ” e x p r e s s { i n p u t } - o { w i l d c a r d s . sample}”
i l s import r e p o r t r u l e r e p o r t : i n p u t : T1=” r e s u l t s . csv ” , F1=” p l o t . pdf ” output : html=” r e p o r t . html ” run : r e p o r t ( ””” ========== Some T i t l e ========== See t a b l e T1 , d i s p l a y some math . . math : : | cq 0 - cq 1 | > {MDIFF} ””” , output . html , ∗∗ i n p u t )
$ snakemake - - summary f i l e date r u l e v e r s i o n s t a t u s plan 500.bam Thu Apr 10 10:55 2015 map 0.7 ok no update 501.bam Thu Apr 10 10:55 2015 map 0.7 ok no update 502.bam Thu Apr 10 10:55 2015 map 0.7 updated i np u t f i l e s update pending 503.bam Thu Apr 09 09:24 2015 map 0.6 v e r s i o n changed to 0.7 no update # update files with changed versions $ snakemake -R `snakemake - - l i s t - v e r s i o n - changes ` # update files with changed code $ snakemake -R `snakemake - - l i s t - code - changes `
Marcel Martin, SciLifeLab Stockholm • Tobias Marschall, Max Planck Institute for Informatics • Sean Davis, NIH • David Koppstein, MIT • Ryan Dale, NIH • Hyeshik Chang, Seoul National University • Karel Brinda, Universit´ e Paris-Est Marne-la-Vall´ ee • Anthony Underwood, Public Health England • Matthew Shirley, Johns Hopkins School of Medicine • Jeremy Leipzig, Childrens Hospital of Philadelphia • Elias Kuthe, TU Dortmund • Paul Moore, Atos SE • Mattias Fr˚ anberg, Karolinska Institute • Kemal Eren • Kyle Meyer • all users and supporters in the mailing list https://bitbucket.org/johanneskoester/snakemake
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}