1300 bp • reference sequence region (usually a region in the genome) Next Generation Sequencing: • short read: tens of millions of sequences, 200 - 300 bp • long read: tens of thousands of sequences, 2.000 - 8.000 bp • unknown origin of reads location 1/18 NGS Data Analysis Course 29-08-2016
1300 bp • reference sequence region (usually a region in the genome) Next Generation Sequencing: • short read: tens of millions of sequences, 200 - 300 bp • long read: tens of thousands of sequences, 2.000 - 8.000 bp • unknown origin of reads location Where do these reads come from? 1/18 NGS Data Analysis Course 29-08-2016
characters: Adenine, Thymine, Guanine, Cytosine. Our sequencing reads become short pieces of strings, our ref- erence sequence becomes a long piece of string. 2/18 NGS Data Analysis Course 29-08-2016
characters: Adenine, Thymine, Guanine, Cytosine. Our sequencing reads become short pieces of strings, our ref- erence sequence becomes a long piece of string. Where can the reads be found within the reference? 2/18 NGS Data Analysis Course 29-08-2016
characters: Adenine, Thymine, Guanine, Cytosine. Our sequencing reads become short pieces of strings, our ref- erence sequence becomes a long piece of string. Where can the reads be found within the reference? in other words, we want to solve a string matching problem 2/18 NGS Data Analysis Course 29-08-2016
regions Reference-related • Our best reference genome still has unknown regions. • Genetic variations exist between organisms. 4/18 NGS Data Analysis Course 29-08-2016
regions Reference-related • Our best reference genome still has unknown regions. • Genetic variations exist between organisms. We need to find approximate instead of exact matches. 4/18 NGS Data Analysis Course 29-08-2016
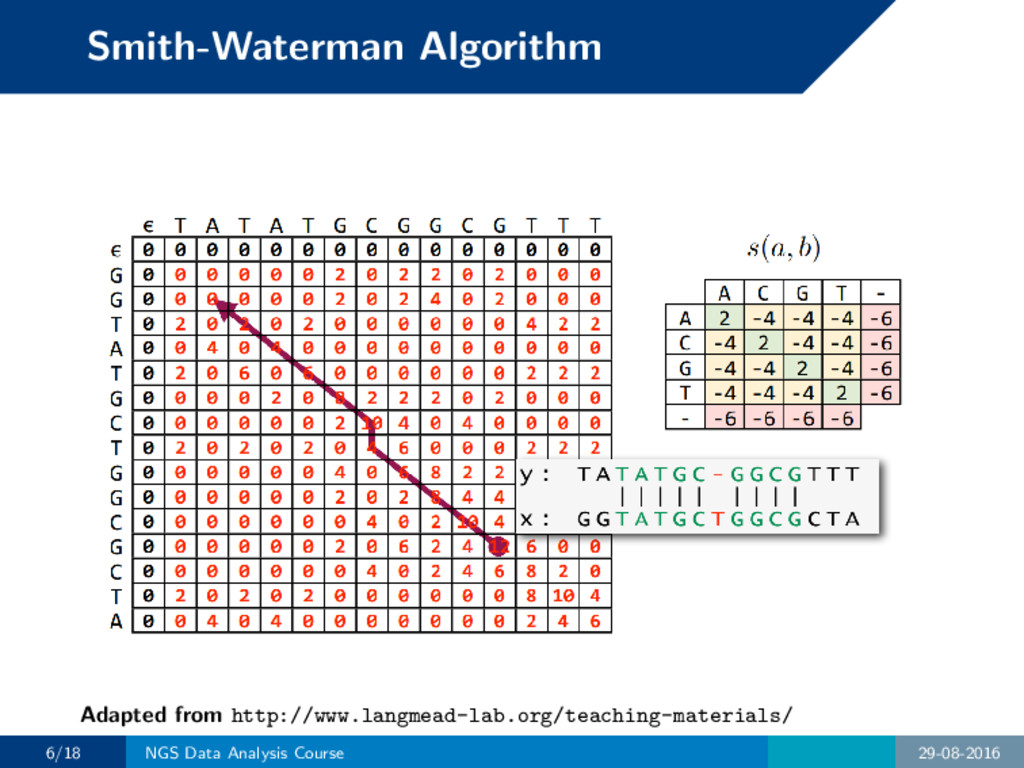
scoring scheme, find the most similar region. • Score for matches, penalize mismatches and gaps. • Strategy: compare optimal alignments of all substrings and pick the highest-scoring one. 5/18 NGS Data Analysis Course 29-08-2016
scoring scheme, find the most similar region. • Score for matches, penalize mismatches and gaps. • Strategy: compare optimal alignments of all substrings and pick the highest-scoring one. Characteristics • Guaranteed to find optimal alignment. • Finds local regions of similarities. • Generalization of Needleman-Wunsch, which finds global similarities. 5/18 NGS Data Analysis Course 29-08-2016
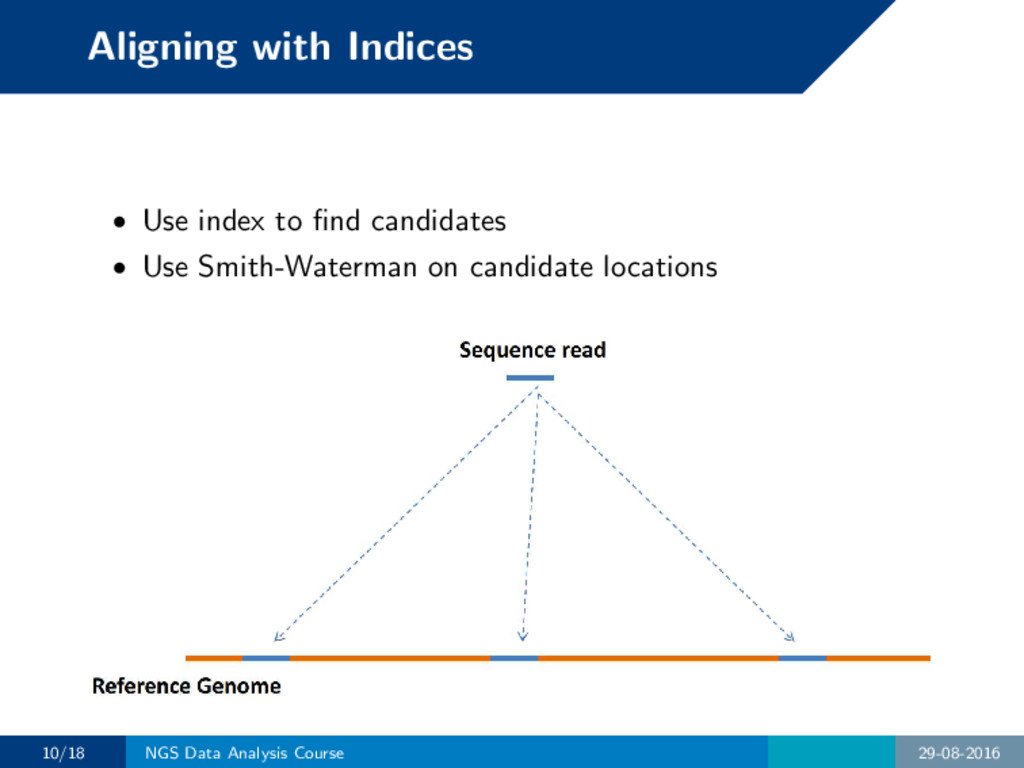
will be huge but most cells will be unused for traceback. • Remember this needs to be done for all reads in both orientations. 8/18 NGS Data Analysis Course 29-08-2016
will be huge but most cells will be unused for traceback. • Remember this needs to be done for all reads in both orientations. How do we reduce space requirement and speed up computa- tion? 8/18 NGS Data Analysis Course 29-08-2016
into another structure more suitable for alignment of sequencing reads. Analogy: book index • Information more or less preserved. • Looking up words become much quicker. 9/18 NGS Data Analysis Course 29-08-2016
into another structure more suitable for alignment of sequencing reads. Analogy: book index • Information more or less preserved. • Looking up words become much quicker. Modern aligners utilize various index data structures: • Hash table • Suffix array • FM index 9/18 NGS Data Analysis Course 29-08-2016
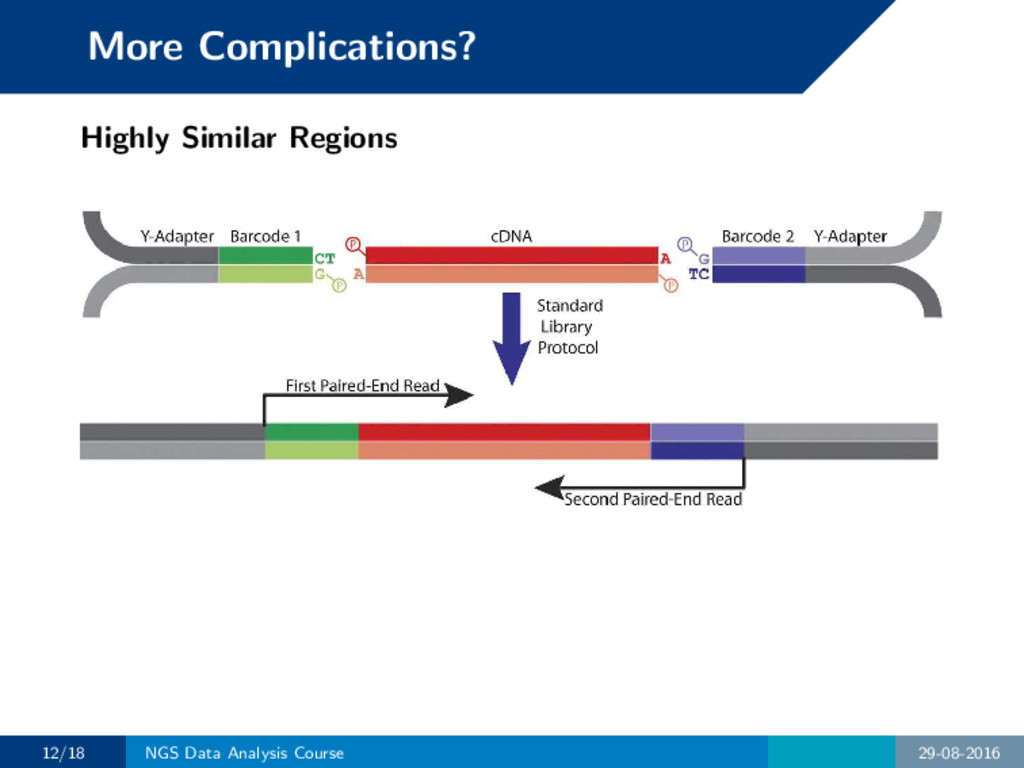
to multiple locations still. How to ensure we map to the correct location? • Use paired-end reads • Use longer reads • Discard them / define a maximum multi alignment limit 11/18 NGS Data Analysis Course 29-08-2016
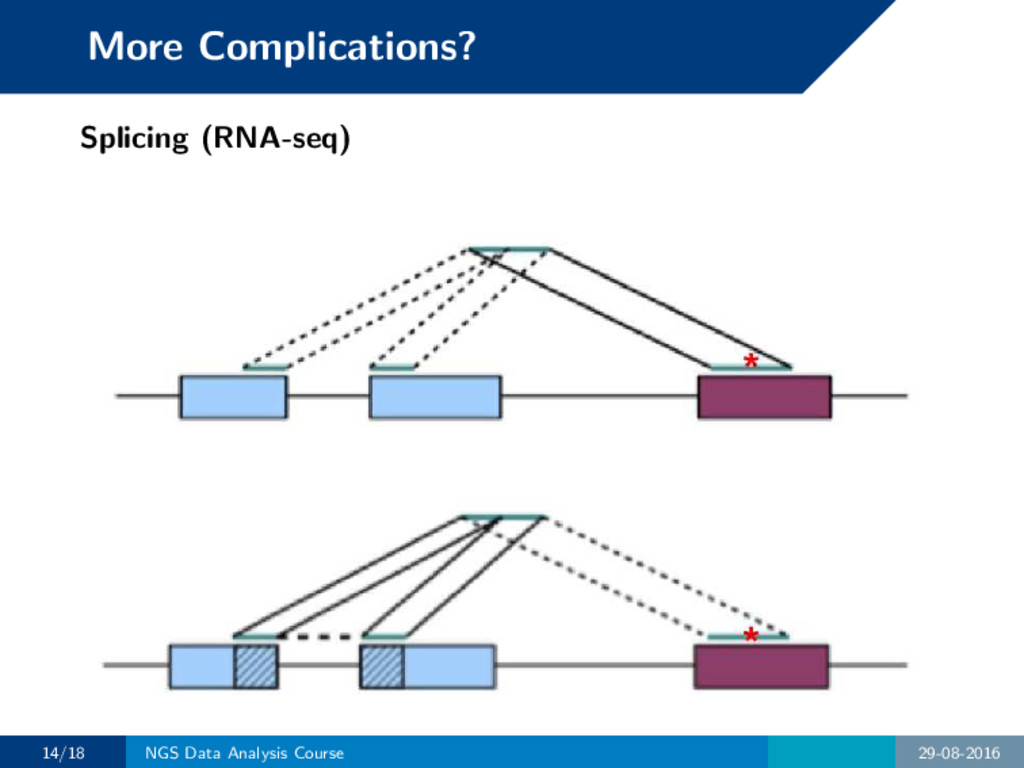
contain introns and our reads span over them. How do we know the correct intron locations? • Align to the transcriptome (at the cost of ignoring novel exons) • Use split read aligners when mapping to the genome 13/18 NGS Data Analysis Course 29-08-2016
always circular. How do we make alignment work with circular references? • Trick: extend reference by adding N bases to the end • Use an aligner that can handle circular references (e.g. GMAP) 15/18 NGS Data Analysis Course 29-08-2016
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}