bias ◦ Sampling bias ◦ Quantifications are relative ◦ Fixed pool of possible reads for a sequencing run ◦ Amount of input material may be different ◦ Read counts are raw data ◦ Different between sequencing runs ◦ Different between different transcripts Disclaimer 4 BioSB RNA-Seq Data Analysis Course 2015
reads across exon-exon junction ◦ Transcriptome ◦ Easier to resolve transcript variants ◦ Restricted to known transcripts ◦ Different mapping challenge: exons used multiple times In practice not so clear cut: ◦ Genome information can be augmented with transcript / splice junction annotations during alignment and/or transcript assembly Choosing the Reference 7 BioSB RNA-Seq Data Analysis Course 2015
Scale ◦ Reads are small but many (millions - hundreds of milions) ◦ One huge reference (MBs ~ GBs) ◦ Mismatches + indels ◦ Technical variation ◦ Population / organism-specific variation ◦ Exon-exon junctions (when aligning to genome)
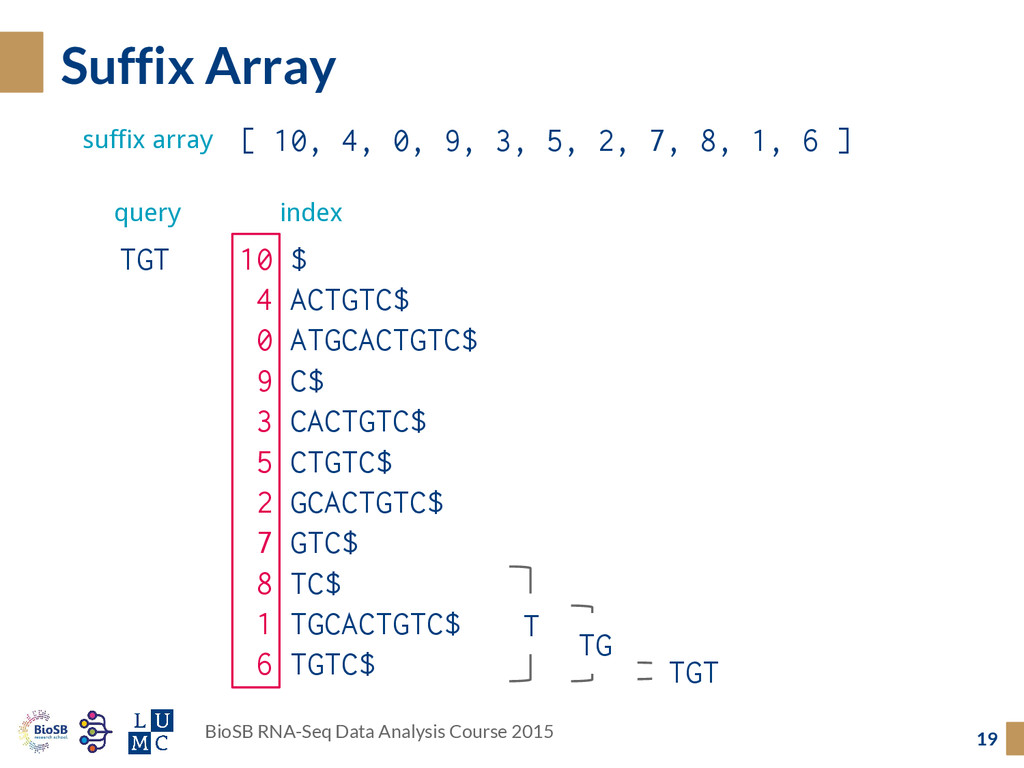
array ◦ Burrows-Wheeler transform The secret sauce is in the details: ◦ How to map over splice junctions? ◦ How to score for mismatches + indels? The (Split) Read Aligners 11 BioSB RNA-Seq Data Analysis Course 2015
into k-mers and compare locations of each to build potential spliced reads ◦ Merges and filters k-mers back into alignments ◦ Novel splice junctions: probabilistic model ◦ Known splice junctions: user input Hash Table-based Aligners 16 BioSB RNA-Seq Data Analysis Course 2015 Publication: Wu (2010) - doi:10.1093/bioinformatics/btq057 Website: http://research-pub.gene.com/gmap/
to split unmapped reads across exons ◦ Maps reads from full exons ◦ Can optionally align to a transcriptome reference first BWT-based Aligners 23 BioSB RNA-Seq Data Analysis Course 2015 Publication: Kim (2013) - doi:10.1186/gb-2013-14-4-r36 Website: https://ccb.jhu.edu/software/tophat/index.shtml
to split unmapped reads across exons ◦ Maps reads from full exons ◦ Can optionally align to a transcriptome reference first HISAT ◦ Main FM index + overlapping indices across reference ◦ Maps spliced reads like STAR, utilizes hierarchical index ◦ Fast + low space requirements BWT-based Aligners 24 BioSB RNA-Seq Data Analysis Course 2015 Publication: Kim (2013) - doi:10.1186/gb-2013-14-4-r36 Website: https://ccb.jhu.edu/software/tophat/index.shtml Publication: Kim (2015) - doi:10.1038/nmeth.3317 Website: https://ccb.jhu.edu/software/hisat/index.shtml
◦ Developer support ◦ Not just one-off publication item ◦ There will be bugs that need fixing Some Considerations 27 BioSB RNA-Seq Data Analysis Course 2015
◦ Developer support ◦ Not just one-off publication item ◦ There will be bugs that need fixing ◦ Community + documentation ◦ Easier to help yourself ◦ Easier to find people who can also help ◦ Higher chance to spot & fix bugs Some Considerations 28 BioSB RNA-Seq Data Analysis Course 2015
◦ Developer support ◦ Not just one-off publication item ◦ There will be bugs that need fixing ◦ Community + documentation ◦ Easier to help yourself ◦ Easier to find people who can also help ◦ Higher chance to spot & fix bugs ◦ Organism annotation status ◦ Less-annotated species should perhaps rely on aligners which are less reliant user-supplied annotation Some Considerations 29 BioSB RNA-Seq Data Analysis Course 2015
◦ Developer support ◦ Not just one-off publication item ◦ There will be bugs that need fixing ◦ Community + documentation ◦ Easier to help yourself ◦ Easier to find people who can also help ◦ Higher chance to spot & fix bugs ◦ Organism annotation status ◦ Less-annotated species should perhaps rely on aligners which are less reliant user-supplied annotation ◦ In-house computing resources ◦ Memory, HDD space, cores, etc. Some Considerations 30 BioSB RNA-Seq Data Analysis Course 2015
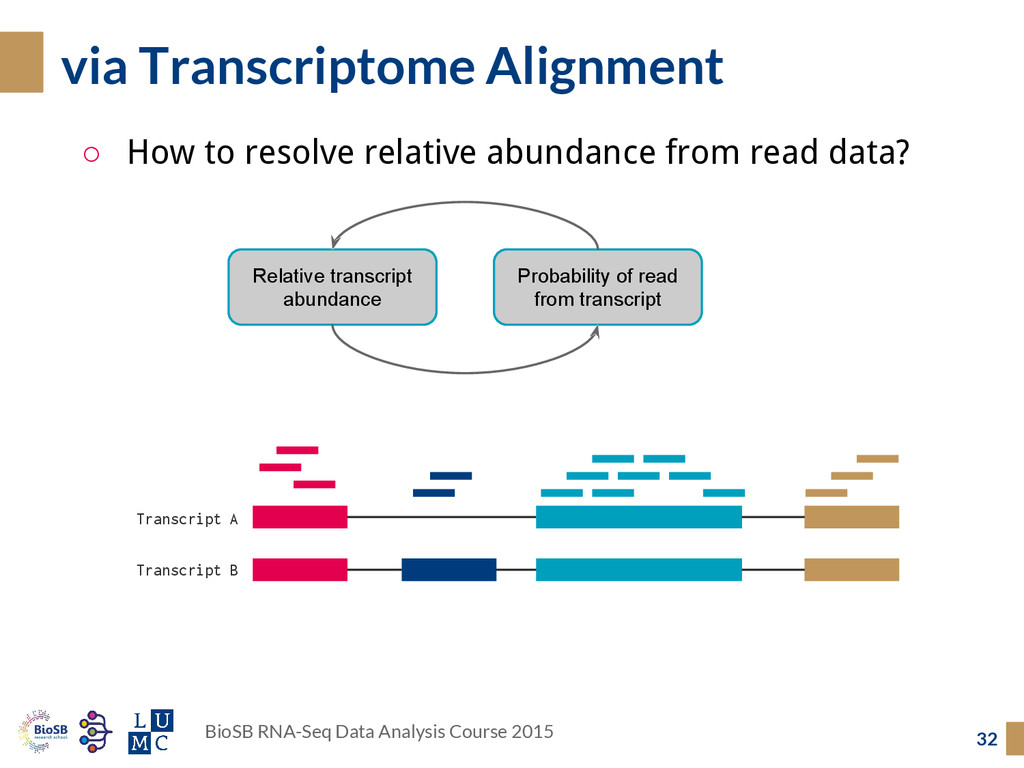
Transcriptome Alignment 32 BioSB RNA-Seq Data Analysis Course 2015 Probability of read from transcript Relative transcript abundance Transcript A Transcript B
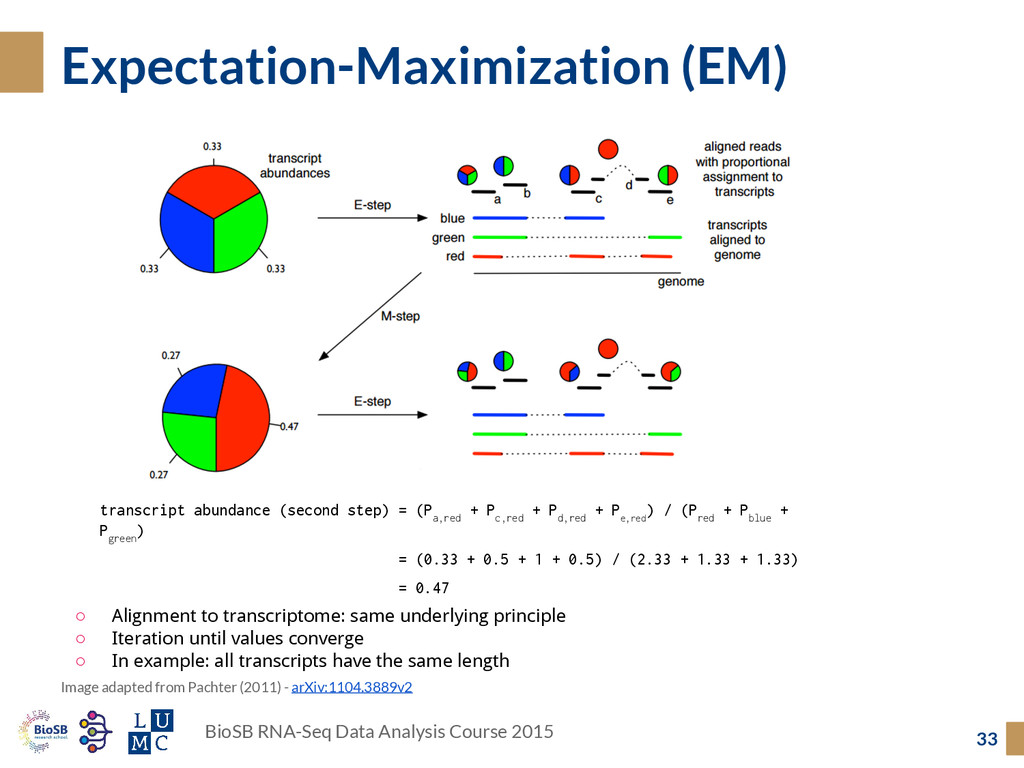
adapted from Pachter (2011) - arXiv:1104.3889v2 transcript abundance (second step) = (P a,red + P c,red + P d,red + P e,red ) / (P red + P blue + P green ) = (0.33 + 0.5 + 1 + 0.5) / (2.33 + 1.33 + 1.33) = 0.47 ◦ Alignment to transcriptome: same underlying principle ◦ Iteration until values converge ◦ In example: all transcripts have the same length
Can visualize counts on transcripts eXpress ◦ Streaming implementation of EM ◦ Can accept results from bowtie as it aligns EM-based Tools 34 BioSB RNA-Seq Data Analysis Course 2015 Publication: Li (2011) - doi:10.1186/1471-2105-12-323 Website: http://deweylab.biostat.wisc.edu/rsem/ Publication: Roberts (2013) - doi:10.1038/nmeth.2251 Website: http://bio.math.berkeley.edu/eXpress/overview.html
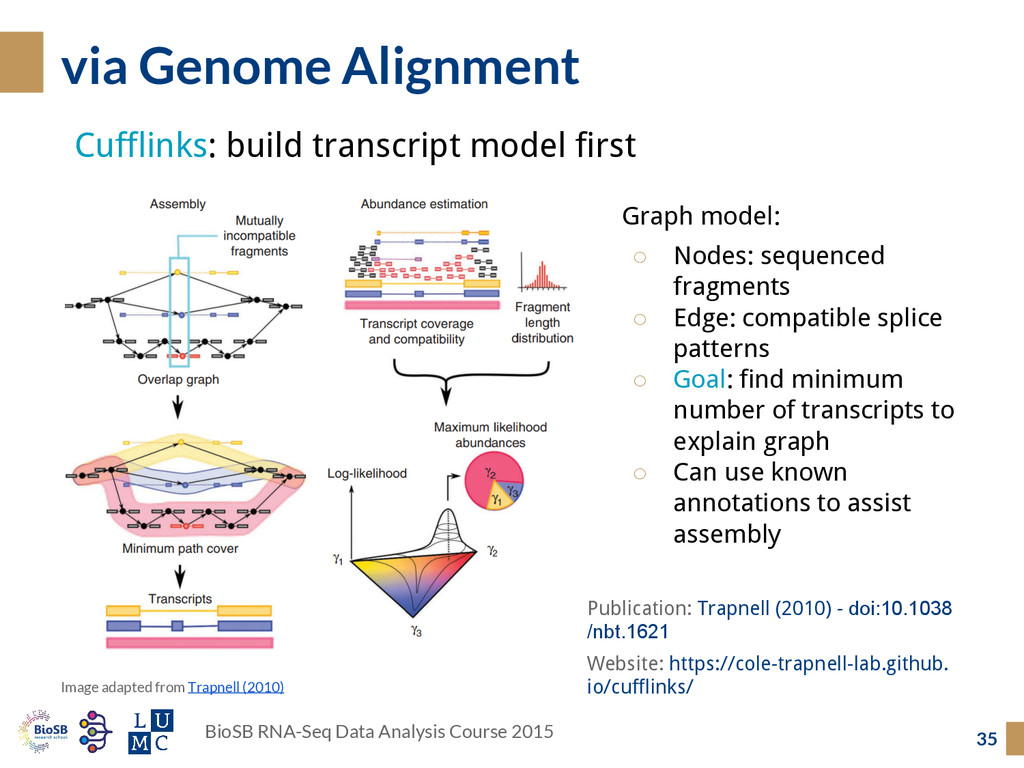
Cufflinks: build transcript model first Image adapted from Trapnell (2010) Graph model: ◦ Nodes: sequenced fragments ◦ Edge: compatible splice patterns ◦ Goal: find minimum number of transcripts to explain graph ◦ Can use known annotations to assist assembly Publication: Trapnell (2010) - doi:10.1038 /nbt.1621 Website: https://cole-trapnell-lab.github. io/cufflinks/
Analysis Course 2015 No abundance estimation - straight to comparisons ◦ Count fragments mapping to features (genes, exons, etc.) ◦ Model distribution feature-wise & compare across samples Image adapted from Finotello (2014) - doi:10.1093/bfgp/elu035
Estimate transcript coverage using k-mer counts in reads and compare to index ◦ From hours to minutes ◦ Limited to known transcripts ◦ Claim: surprisingly comparable
Estimate transcript coverage using k-mer counts in reads and compare to index ◦ From hours to minutes ◦ Limited to known transcripts ◦ Claim: surprisingly comparable Kallisto ◦ de Bruijn graphs for index ◦ Claims higher accuracy than Sailfish Sailfish ◦ First paper with this approach ◦ k-mer index + aux. arrays Publication: Patro (2014) - doi:10.1038/nbt.2862 Website: https://www.cs.cmu. edu/~ckingsf/software/sailfish Publication: Bray (2015) - arXiv:1505.02710v2 Website: https://github.com/pachterlab/kallisto
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}