Team lcolladotor Journal Club Presentation: "Cell-type deconvolution methods for spatial transcriptomics"
doi: https://doi.org/10.1038/s41576-025-00845-y
Recording link to our journal club presentation: https://www.youtube.com/watch?v=xDjFJRGp56o
Presented By: Manisha Barse
Date: August 13, 2025
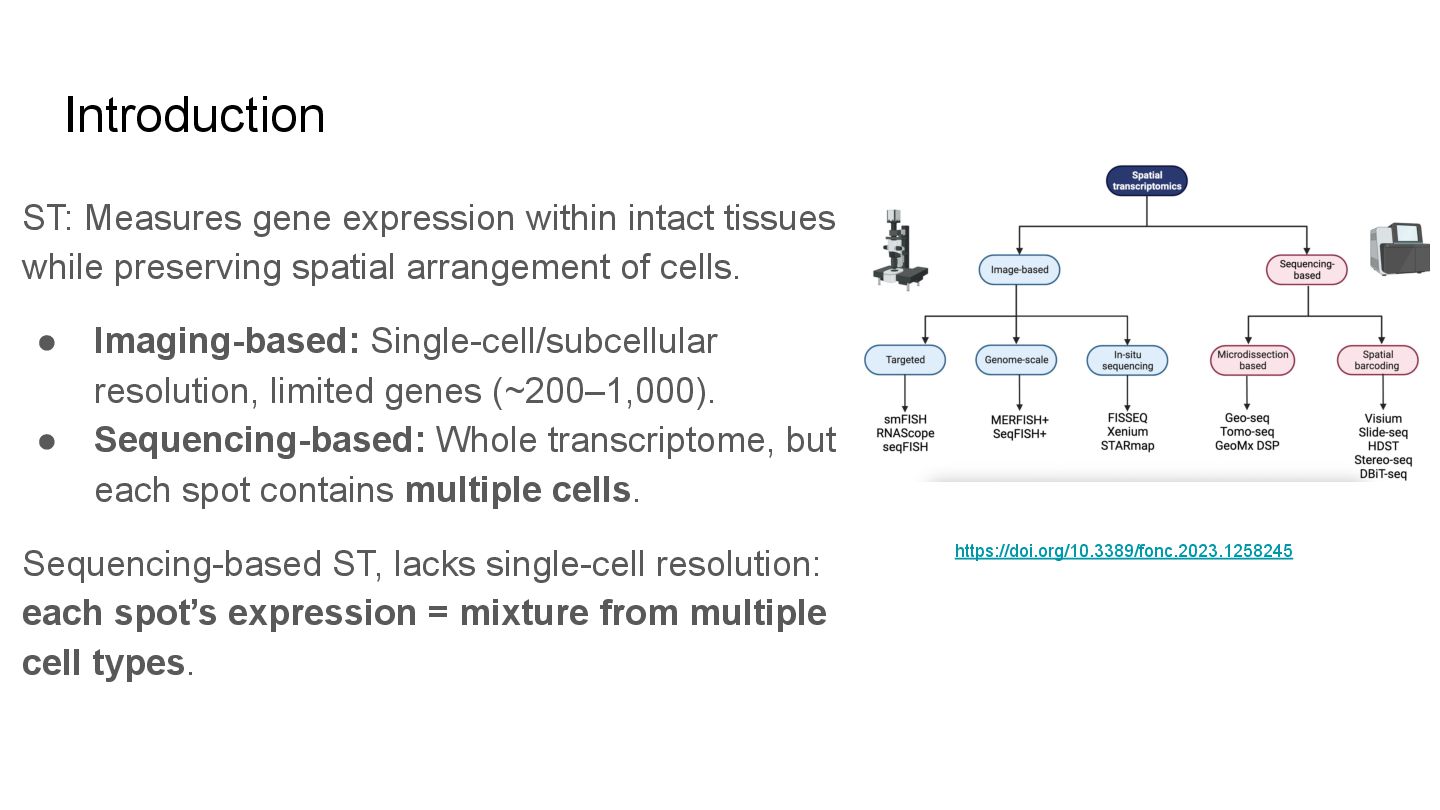
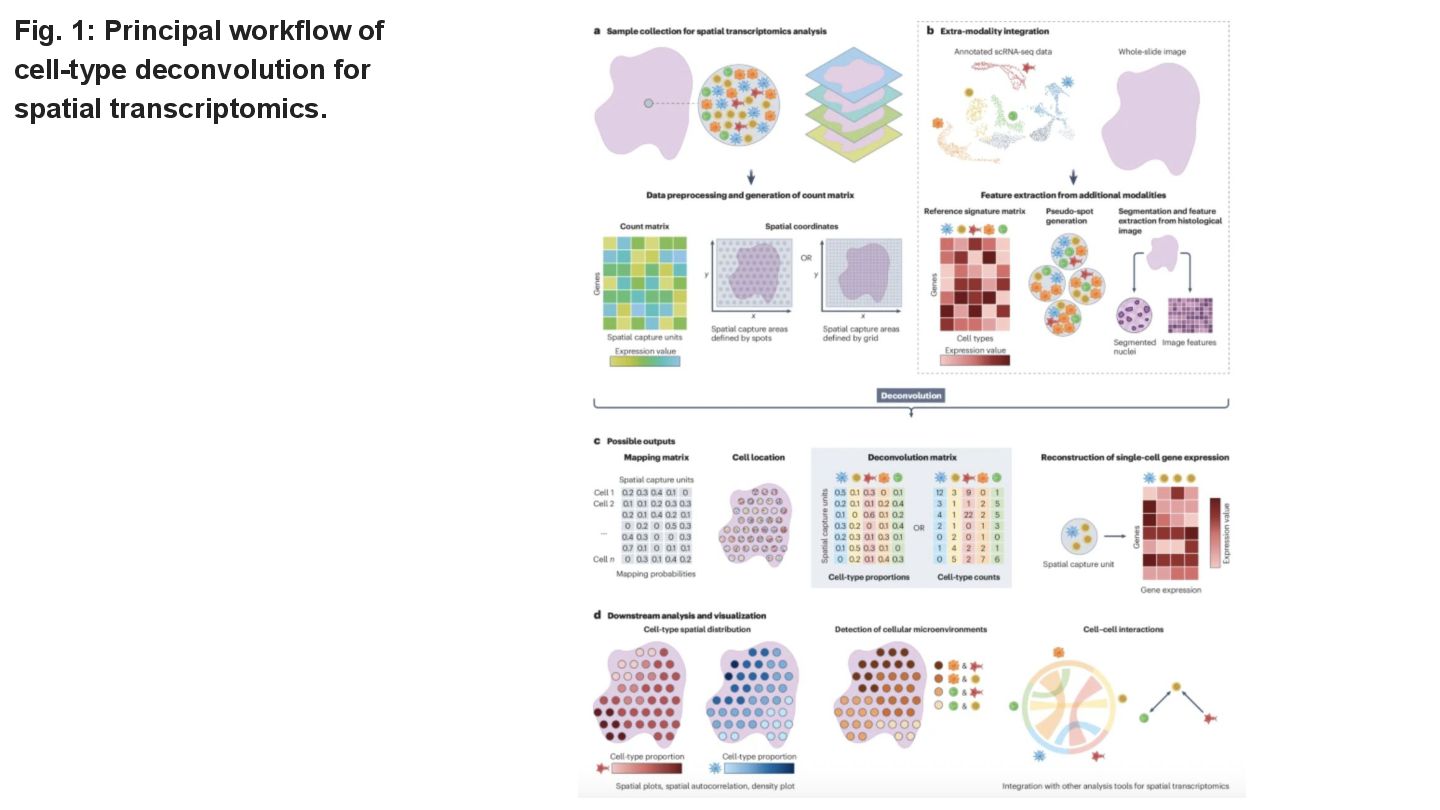
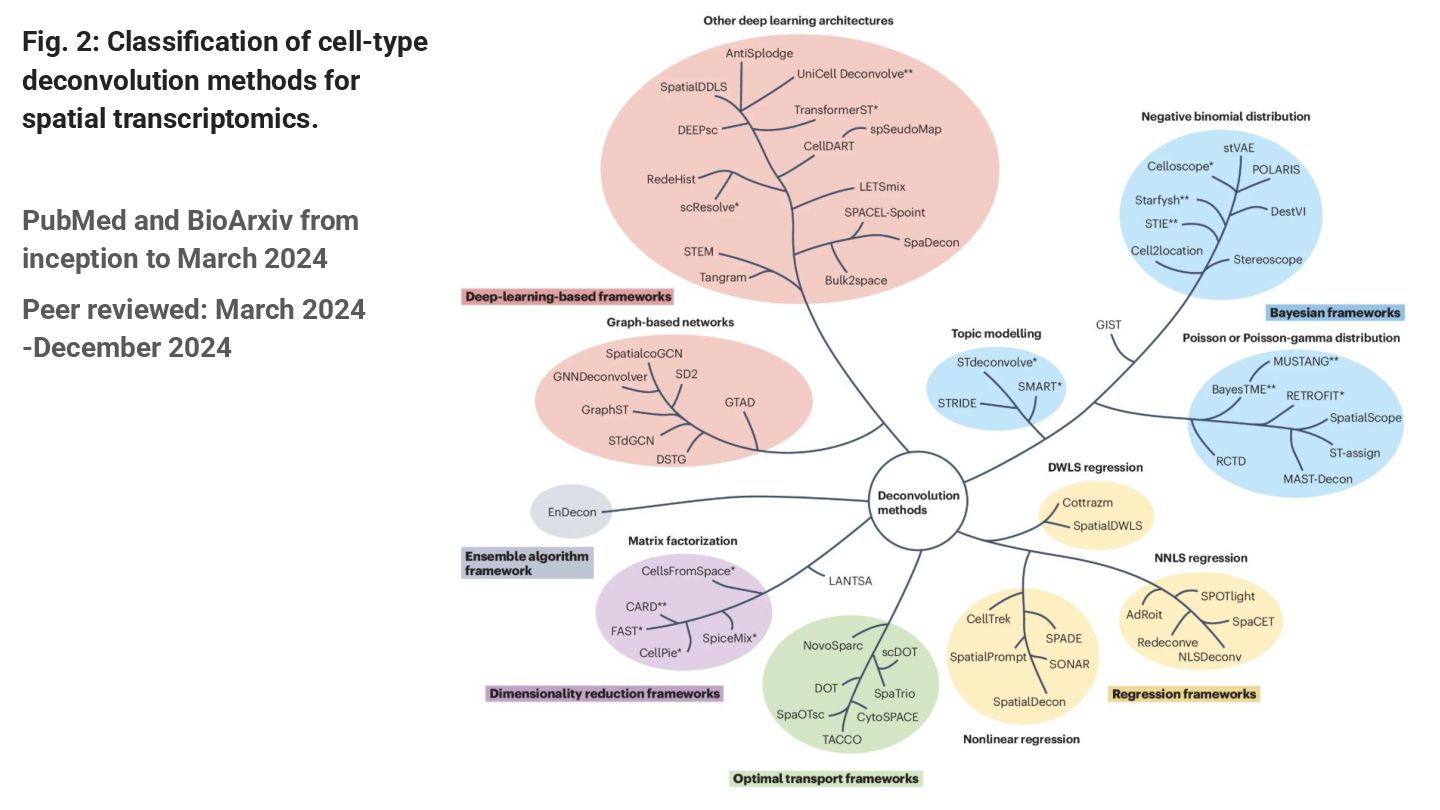
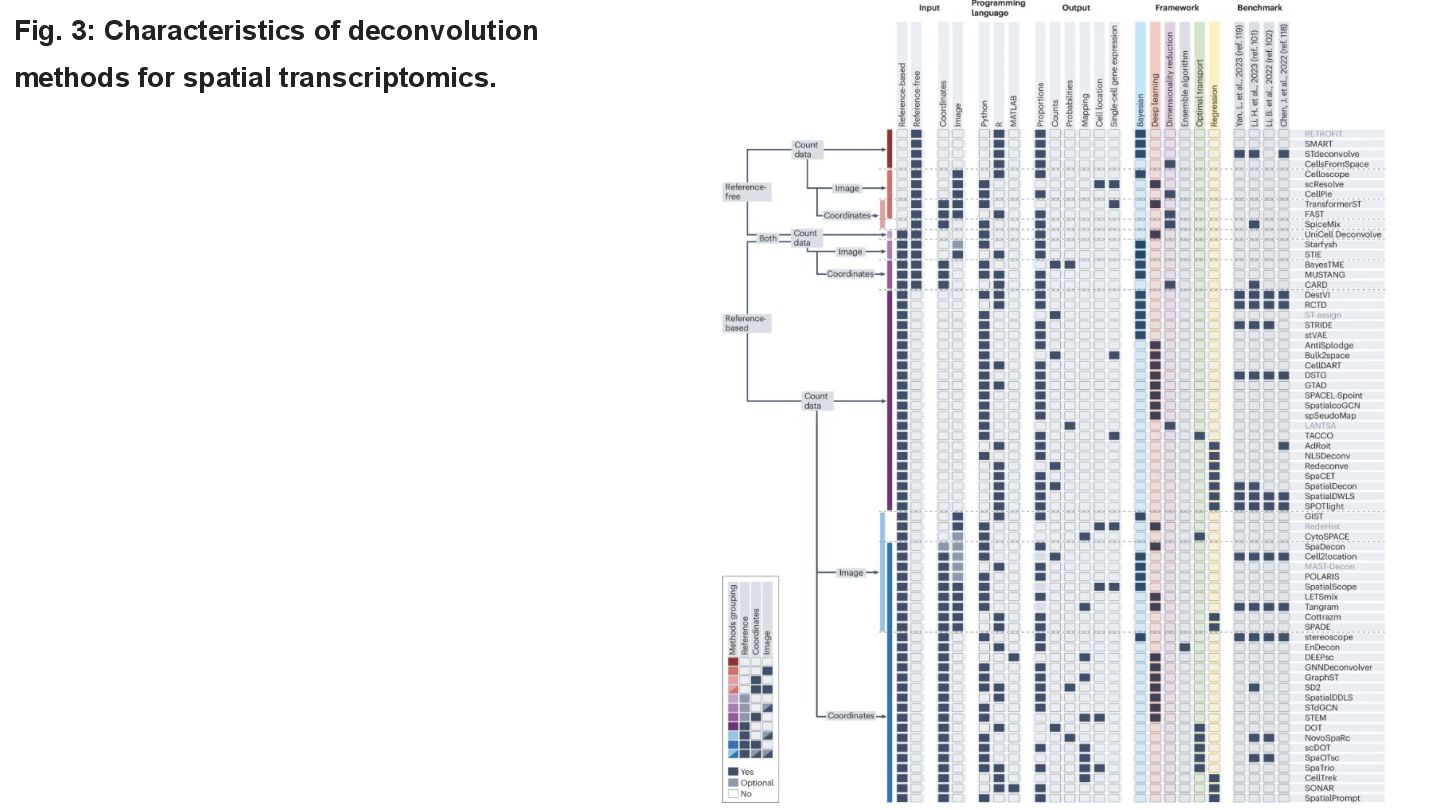
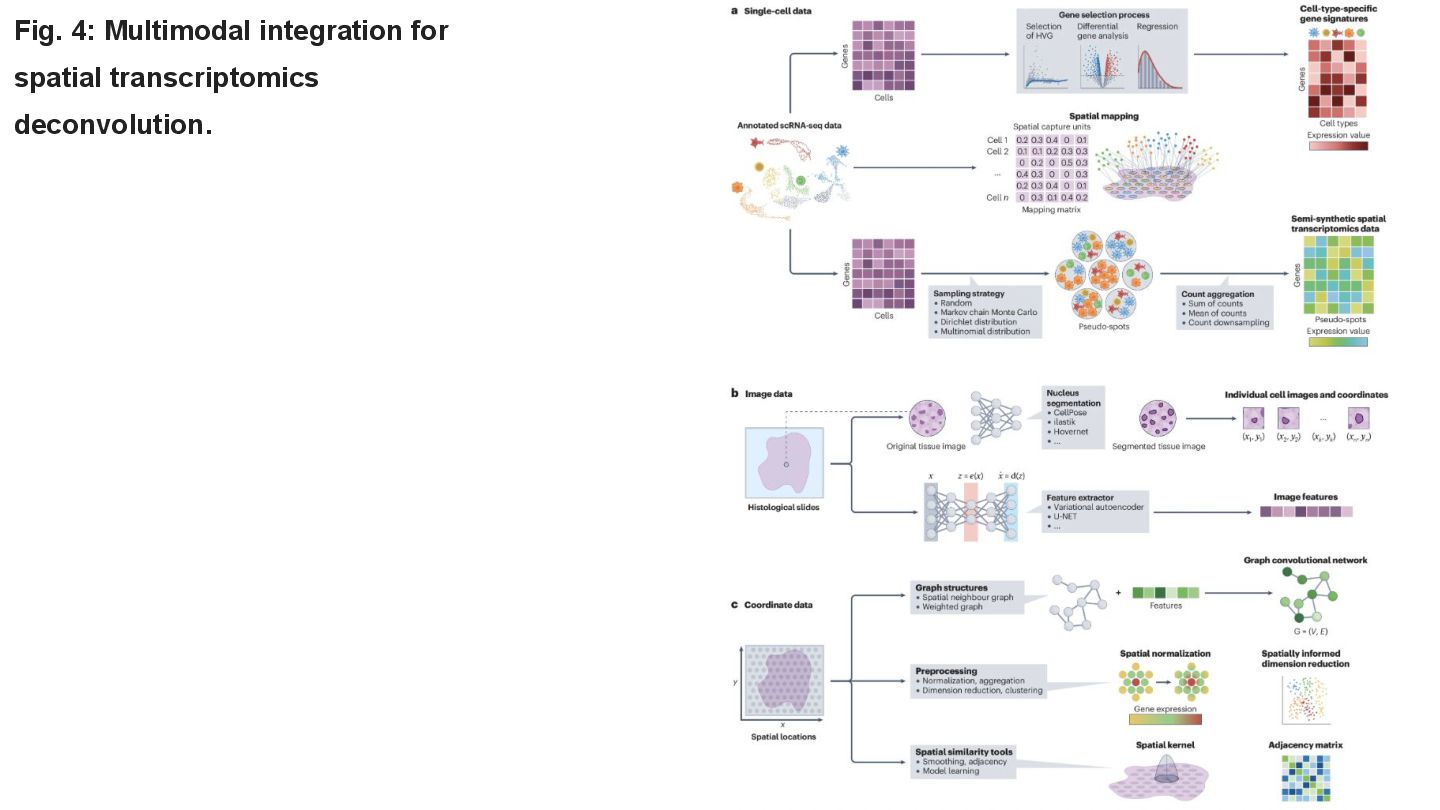
This paper reviews the latest computational approaches for cell-type deconvolution in spatial transcriptomics data. It categorizes existing tools, compares their features, and highlights how they integrate different data modalities, along with a useful interactive tool to compare them.
#transcriptomics #spot-deconvolution #benchmarks #spatial_transcriptomics
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}