l’analyse de données RNA-seq 42 Principales problématiques • Si la profondeur de séquençage est suffisante : possible d’inférer directement la structure complète du transcriptome (TSS, TTS, jonctions d’épissage, transcrits …). Mais CE N’EST PAS NOTRE OBJECTIF ICI à Utilisation d’un fichier GTF pour guider l’alignement • La référence utilisée correspond à un mélange hétérogènes d’haplotypes à Autoriser des mis-matches • Les séquences à aligner contiennent des erreurs de séquençage à Autoriser des mis-matches • Les séquences correspondent, en majorité, à des mRNA matures à Tenir compte de l’épissage
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
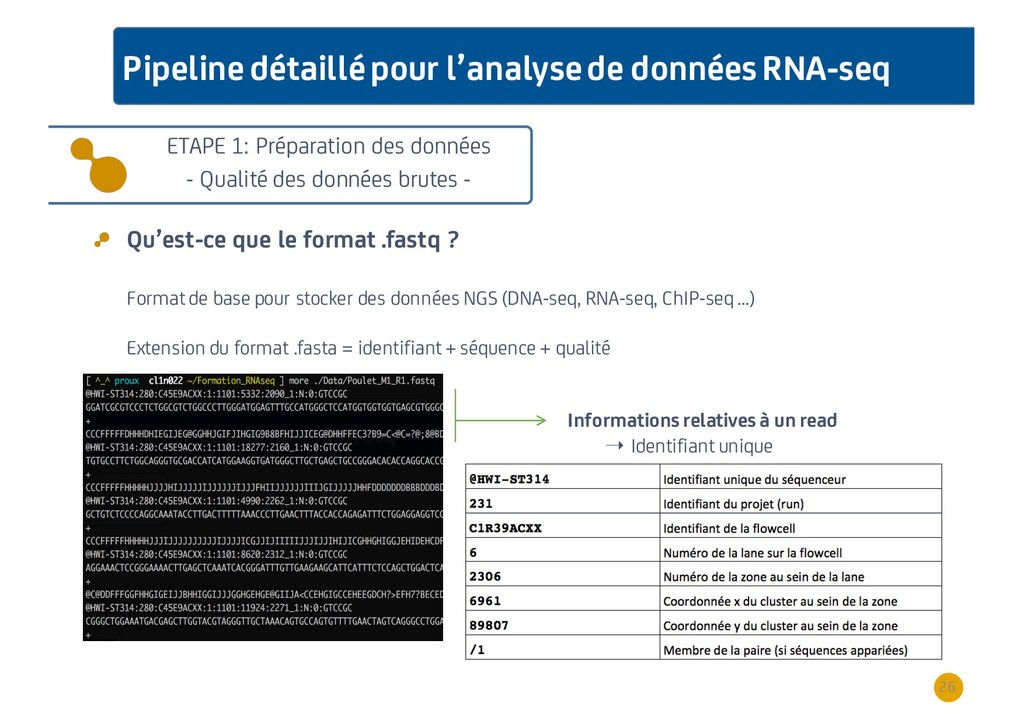{kind=link}
{kind=link}
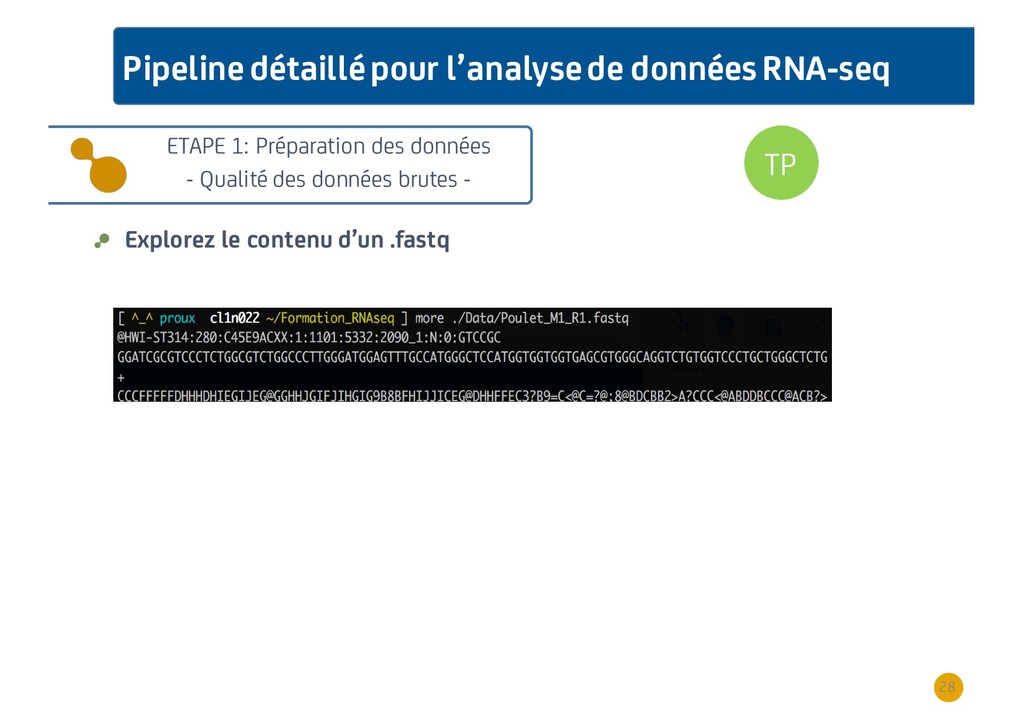{kind=link}
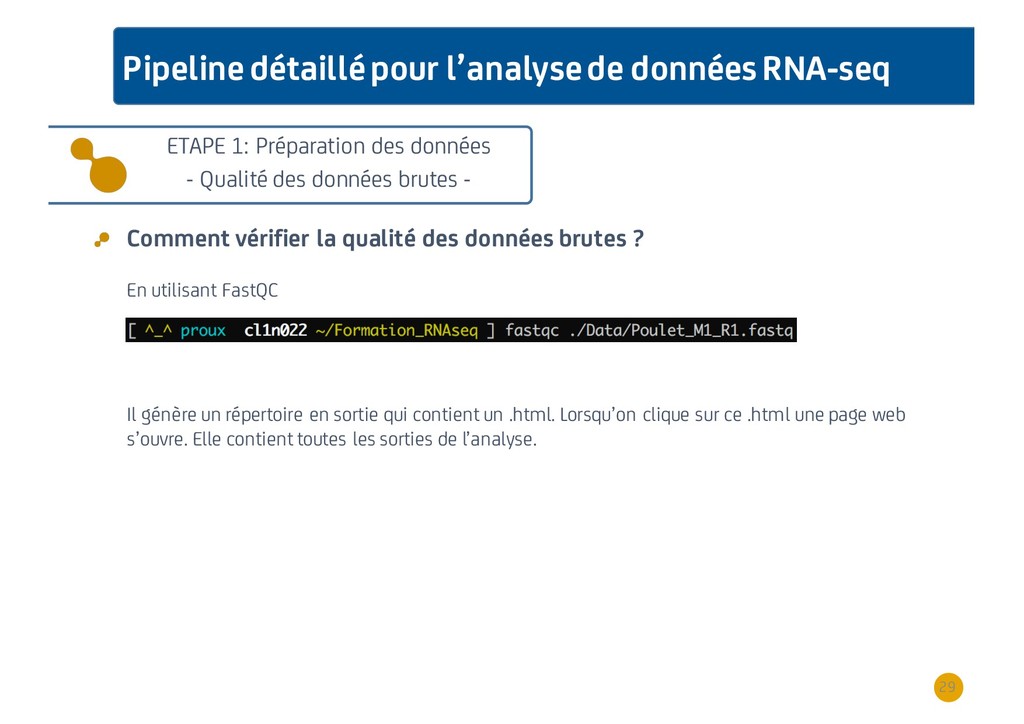{kind=link}
{kind=link}
{kind=link}
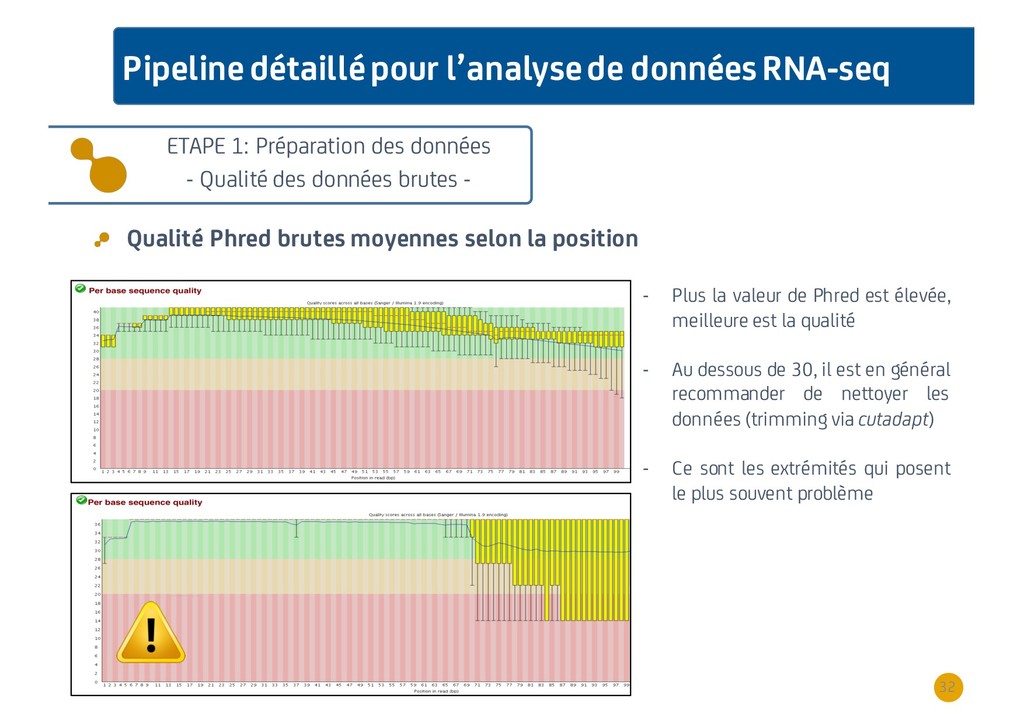{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
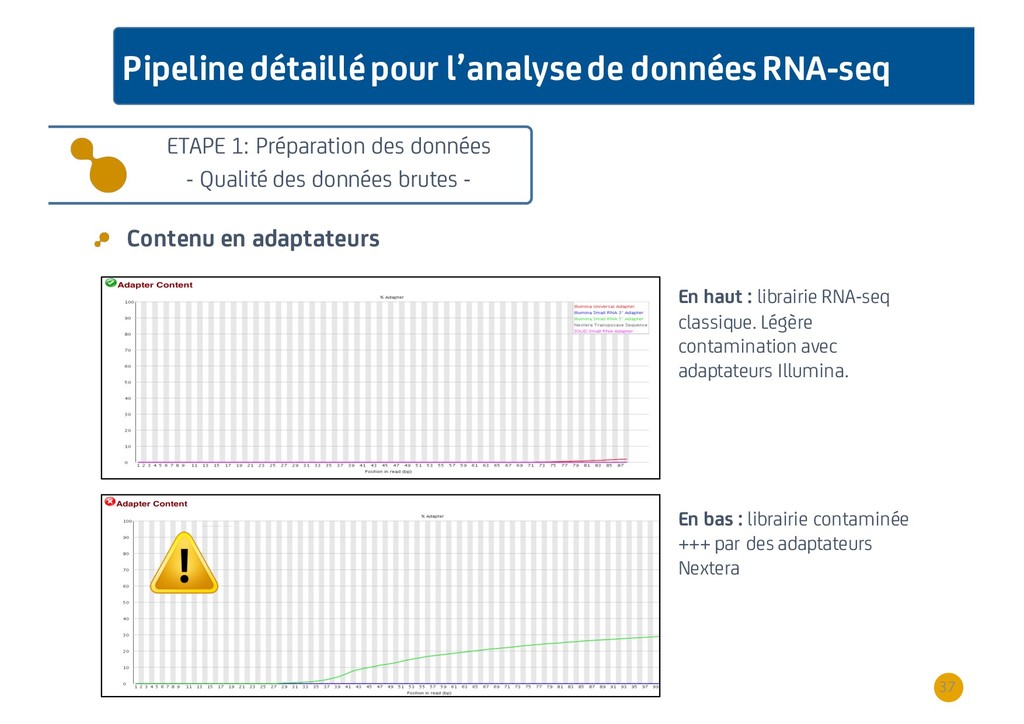{kind=link}
{kind=link}
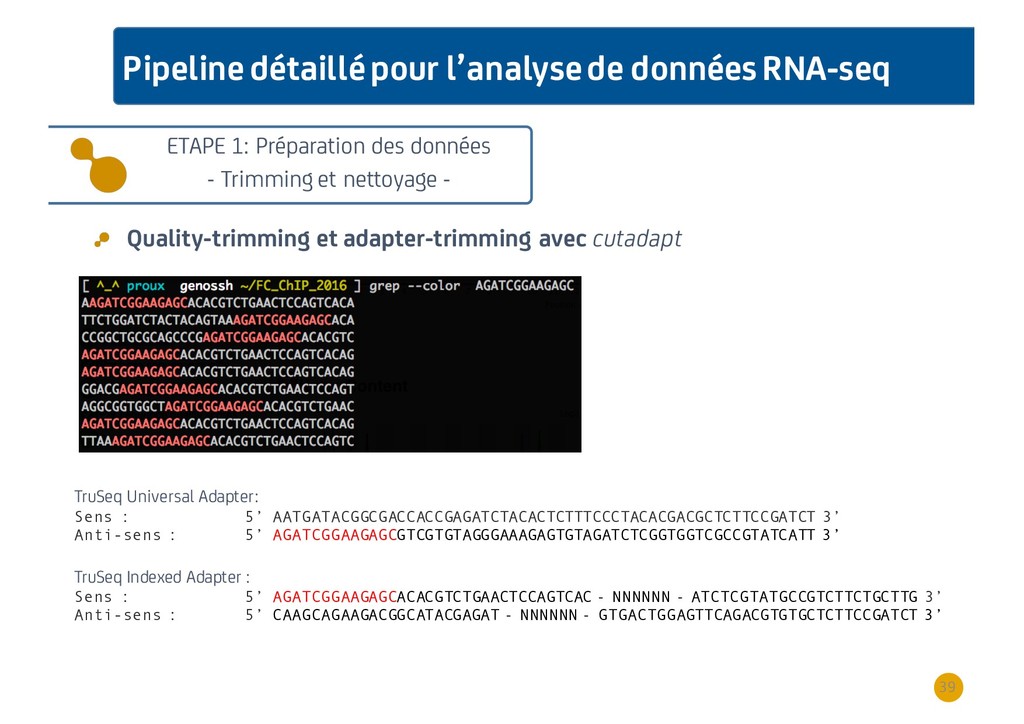{kind=link}
{kind=link}
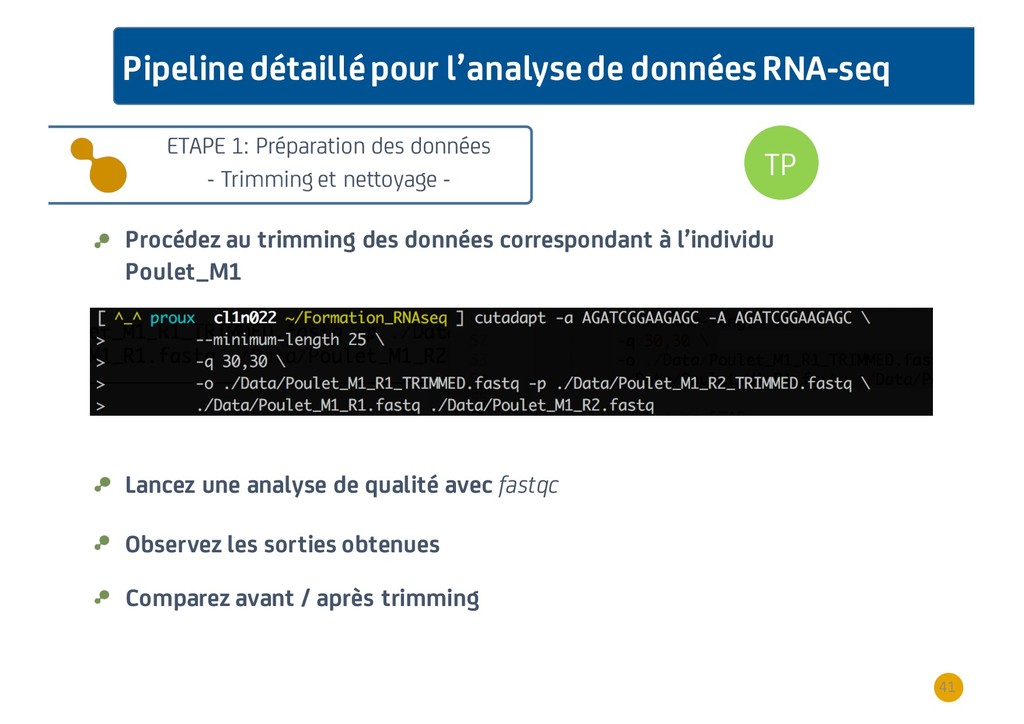{kind=link}
{kind=link}
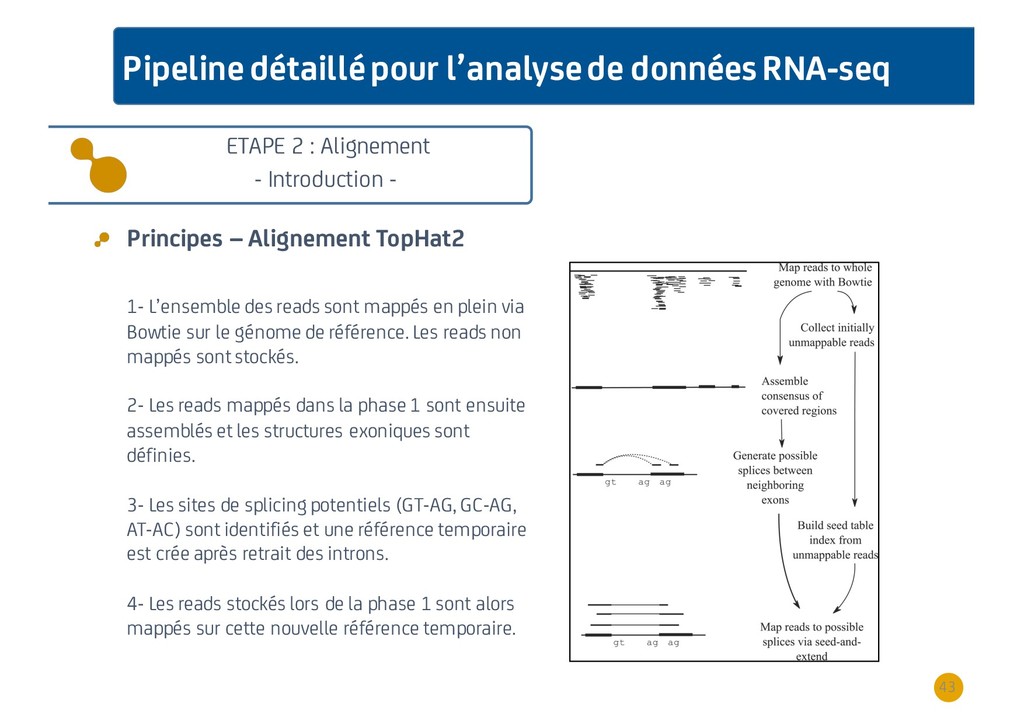{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
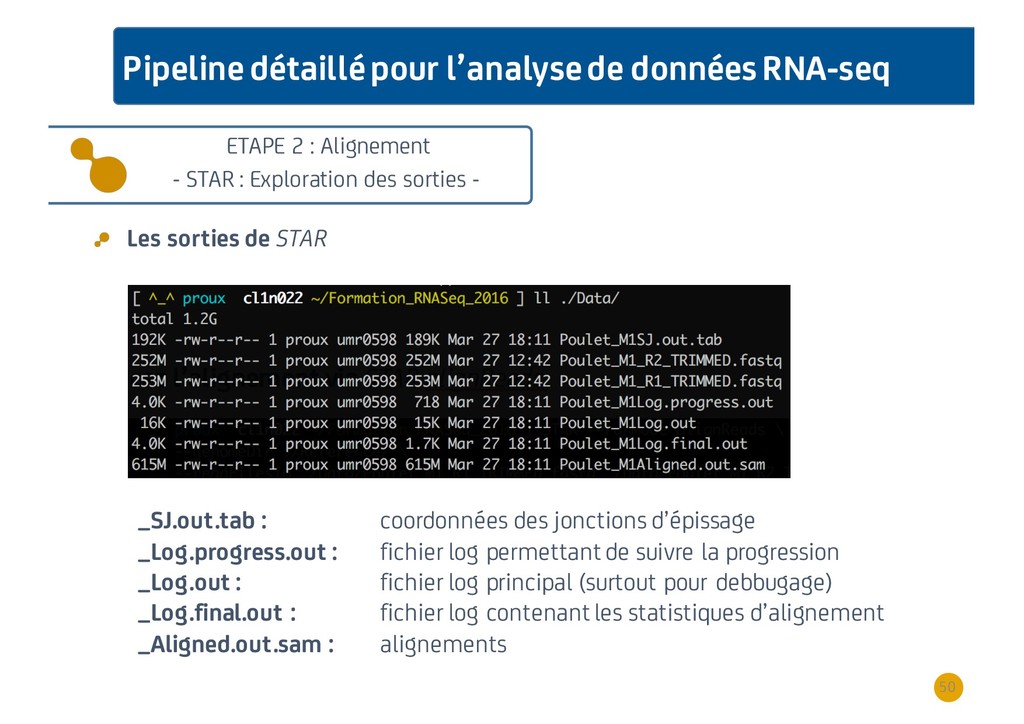{kind=link}
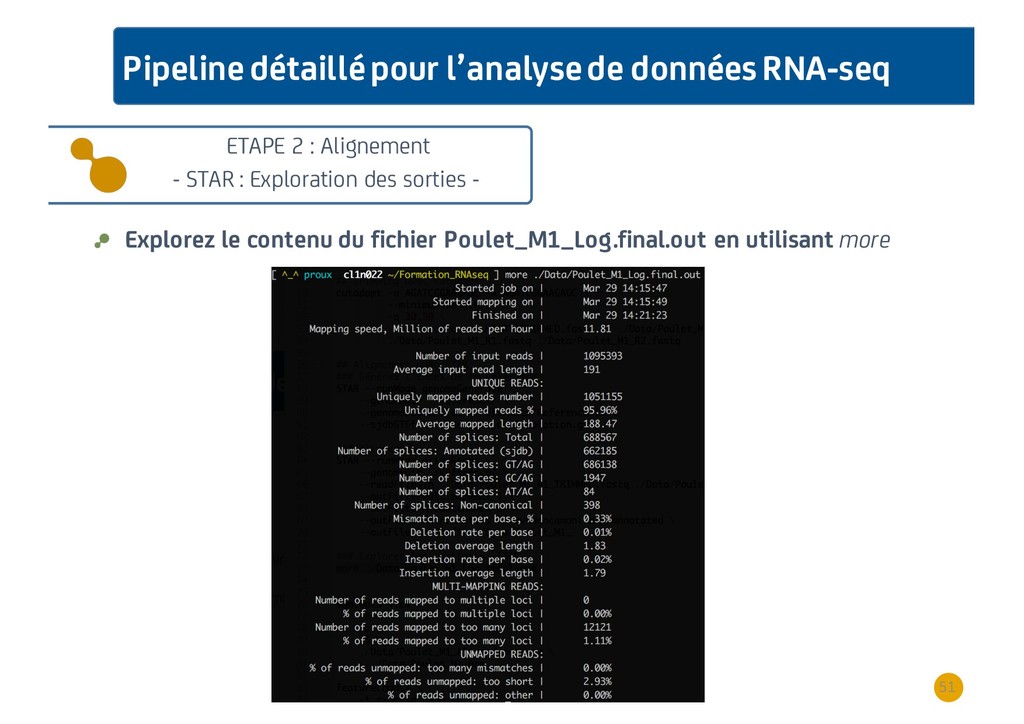{kind=link}
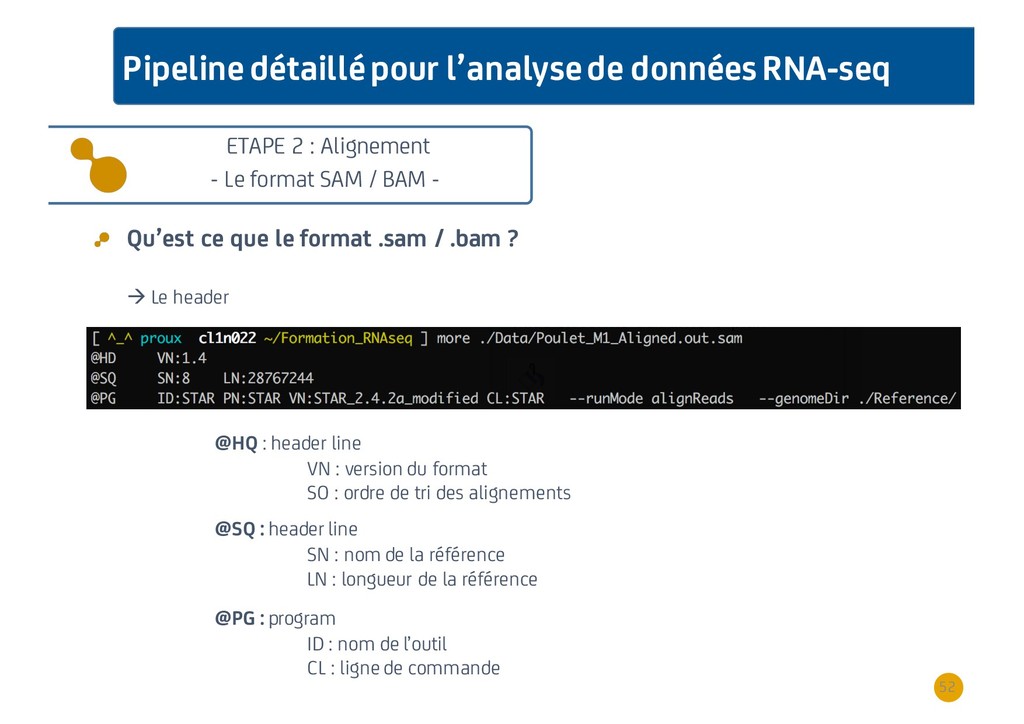{kind=link}
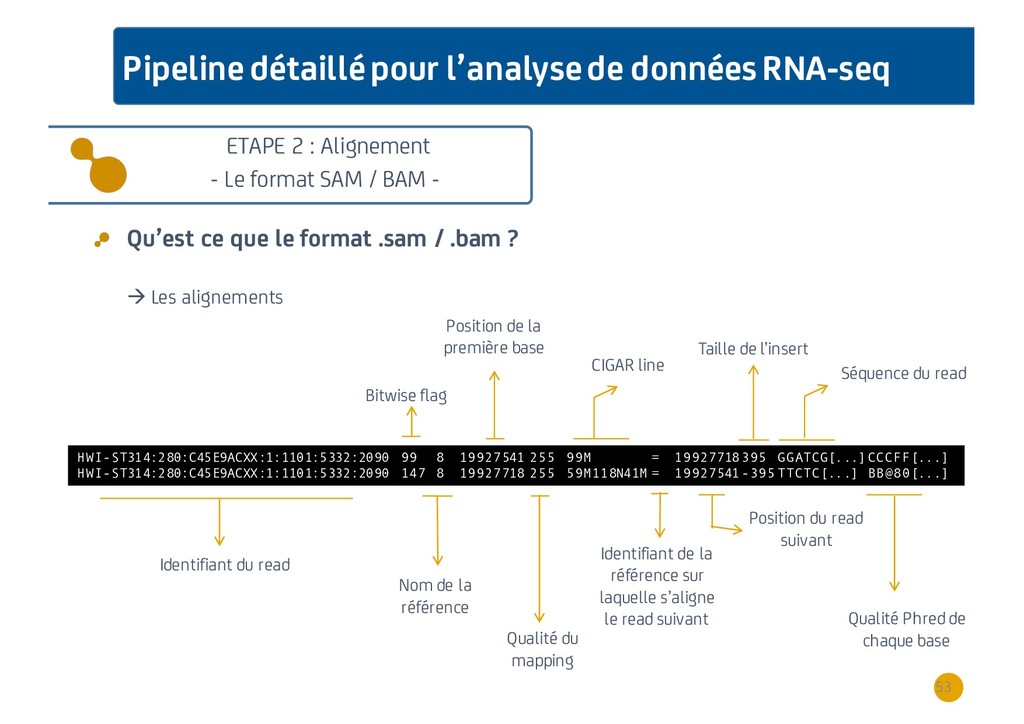{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
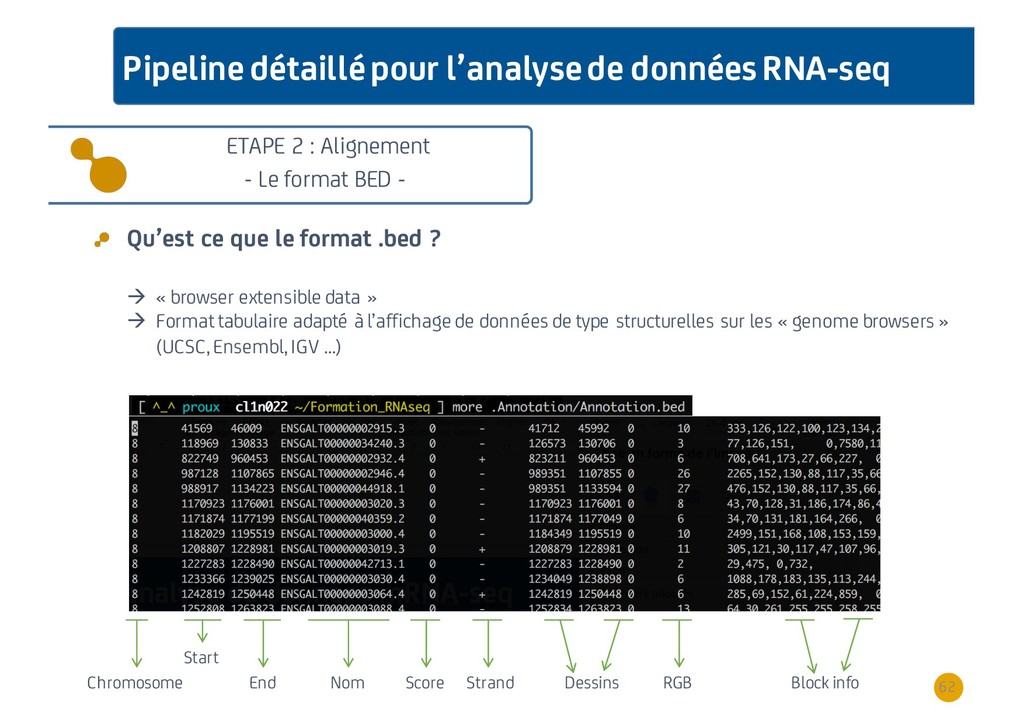{kind=link}
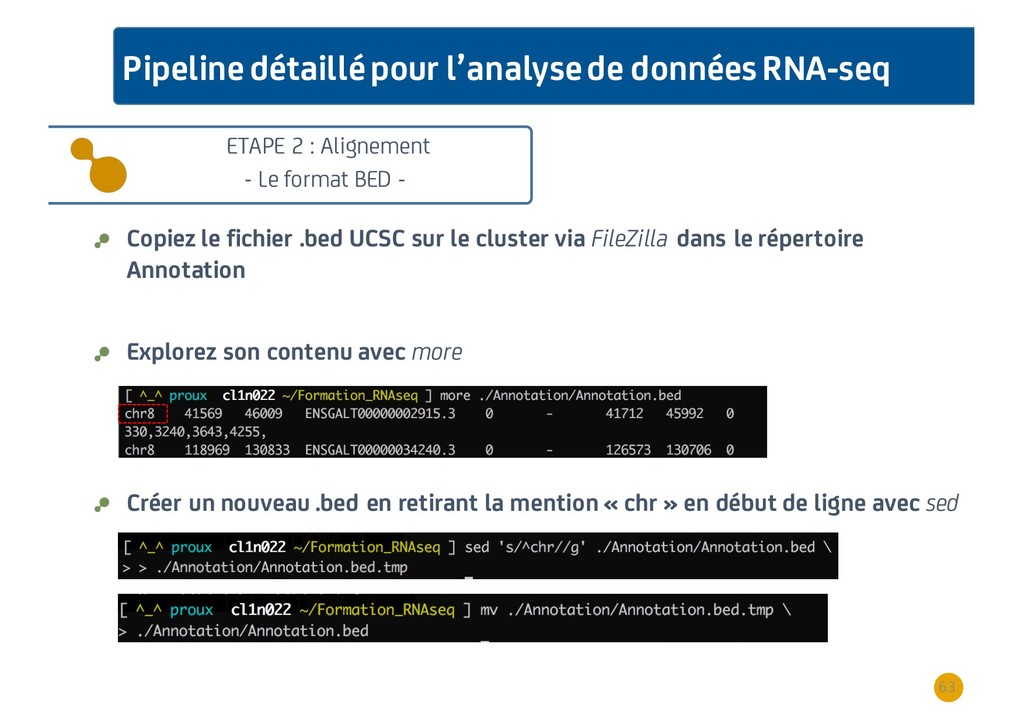{kind=link}
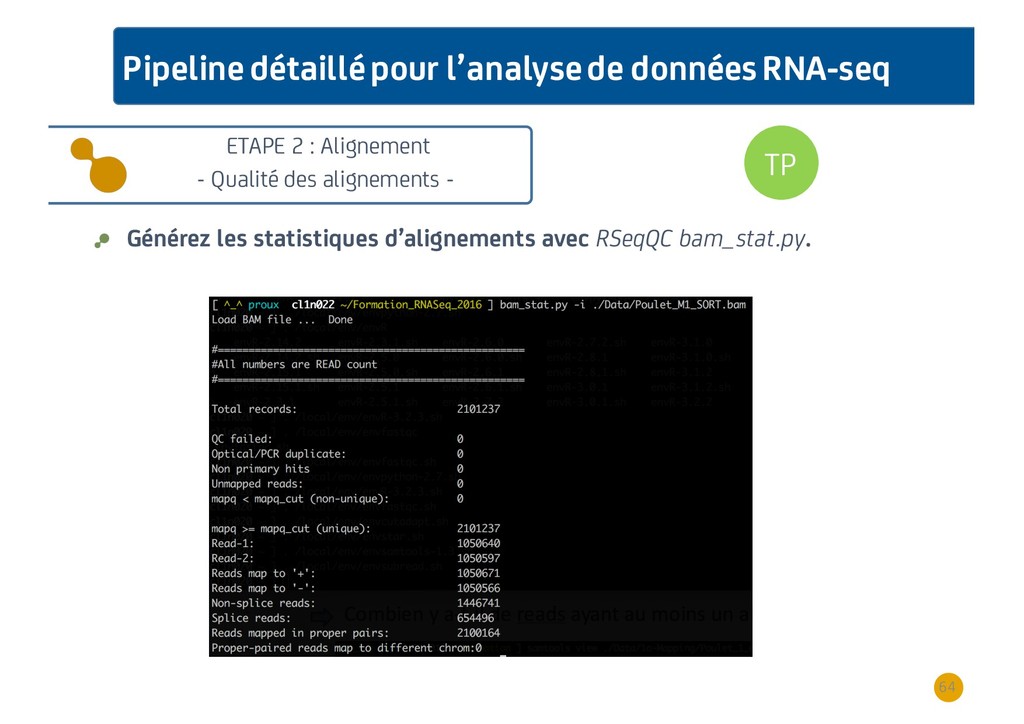{kind=link}
{kind=link}
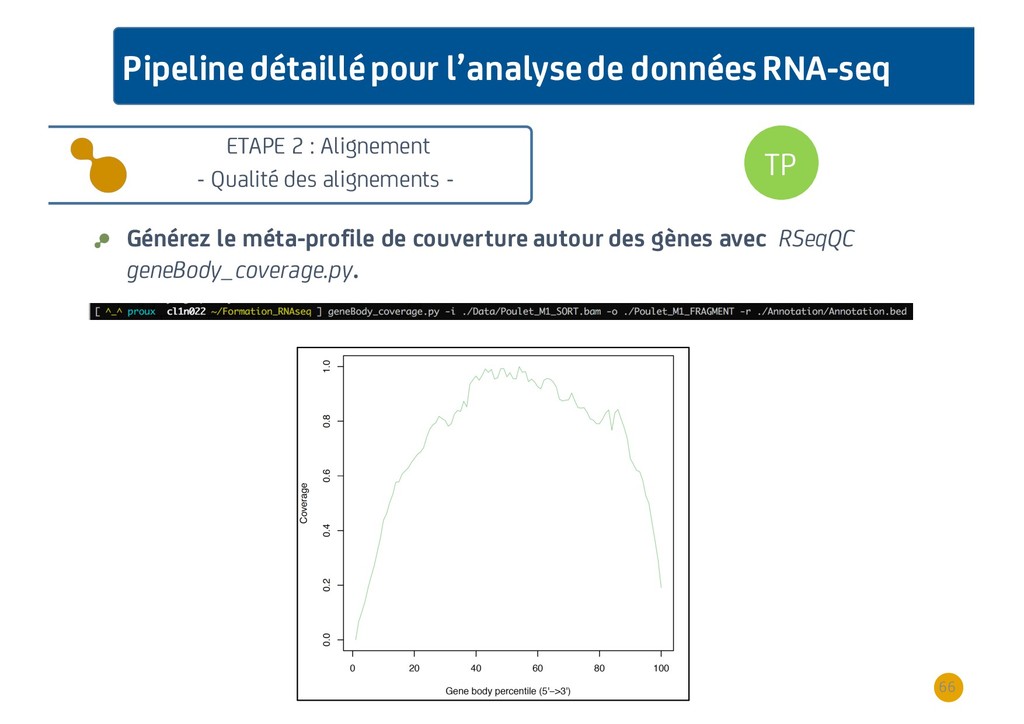{kind=link}
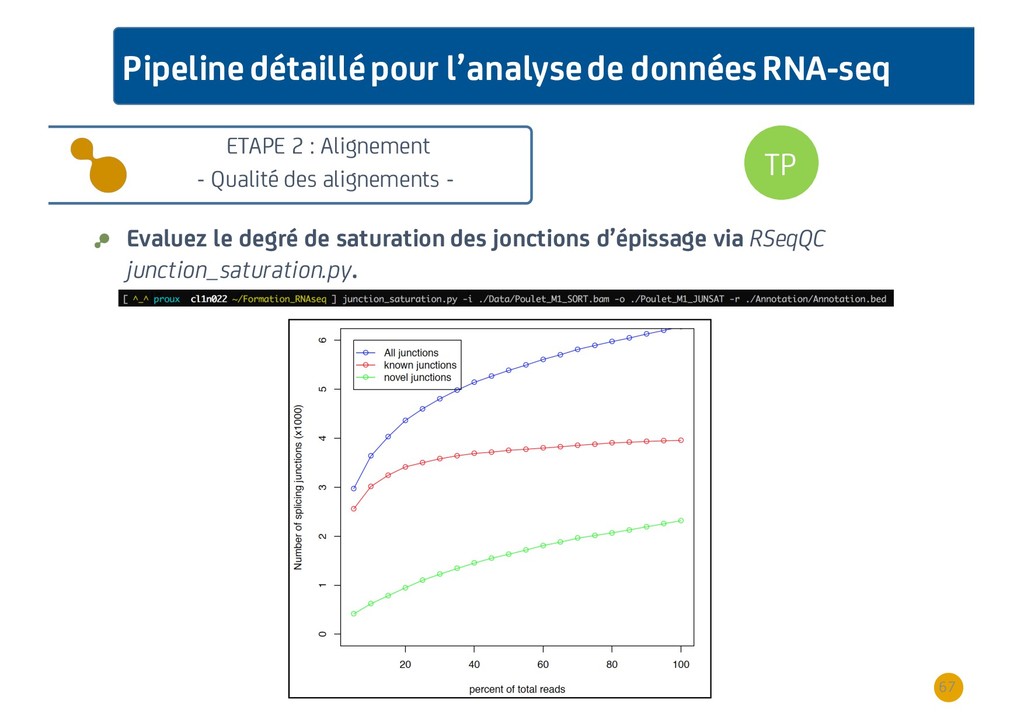{kind=link}
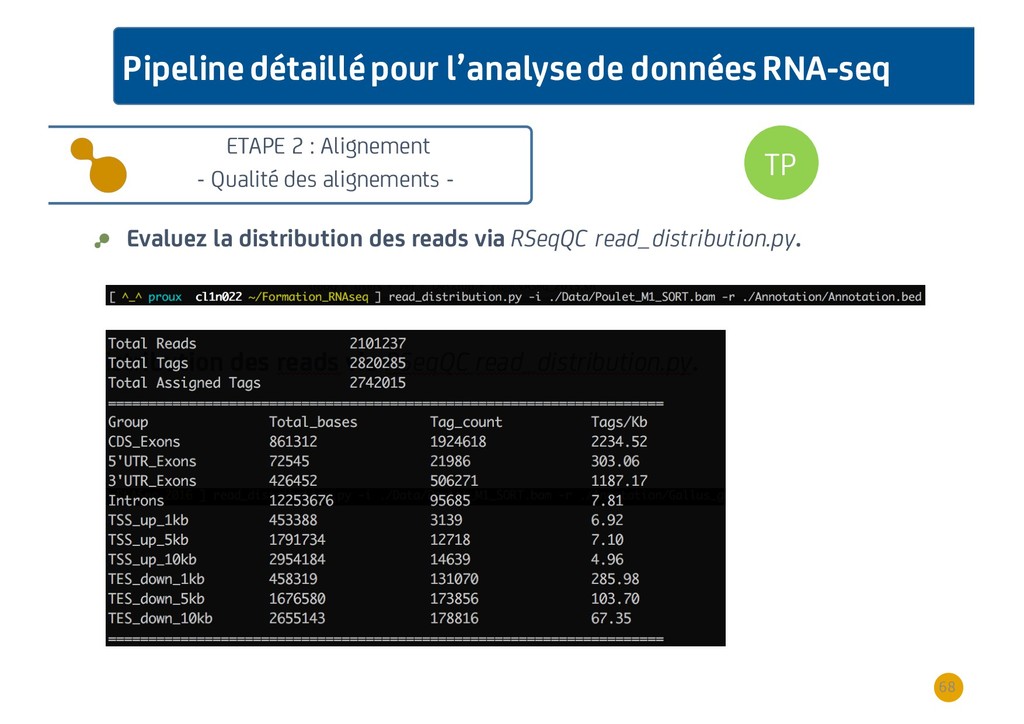{kind=link}
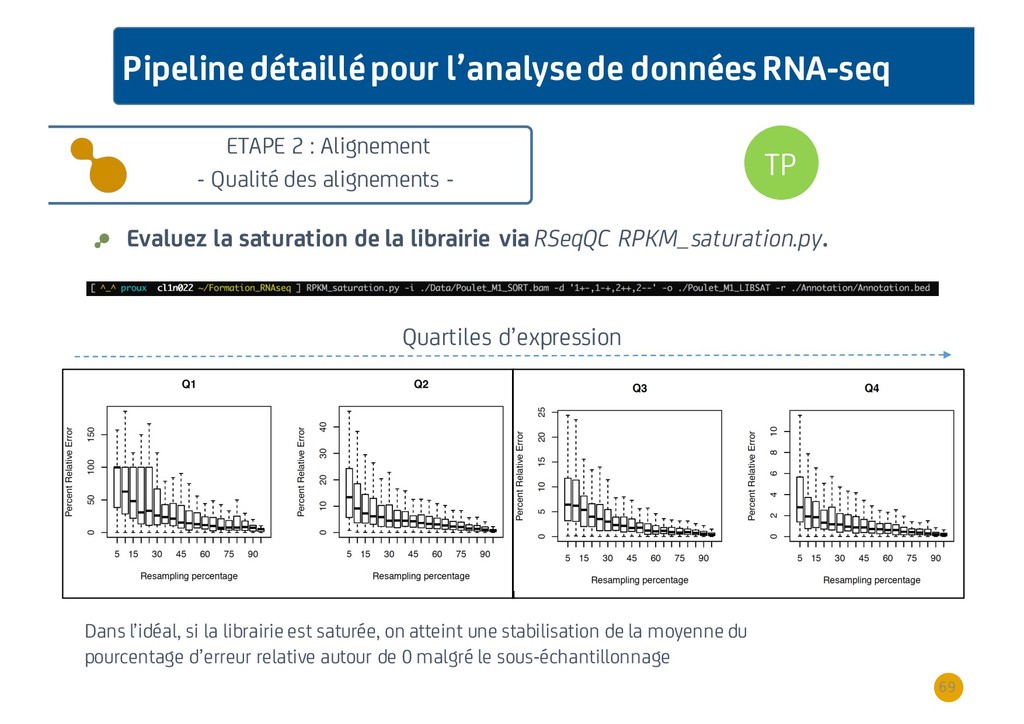{kind=link}
{kind=link}
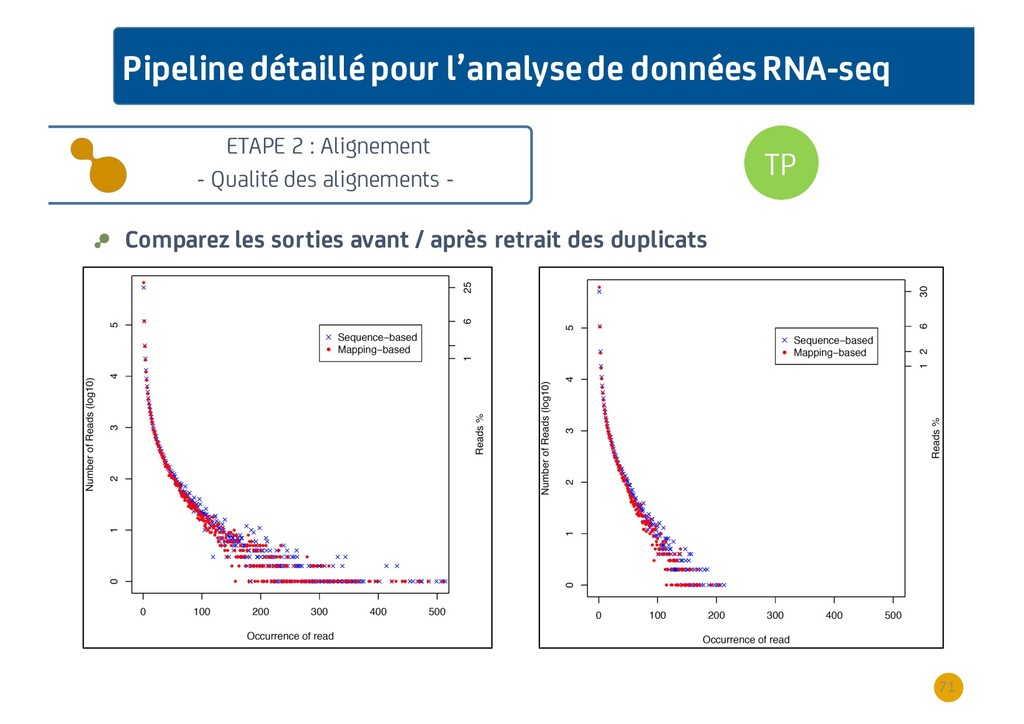{kind=link}
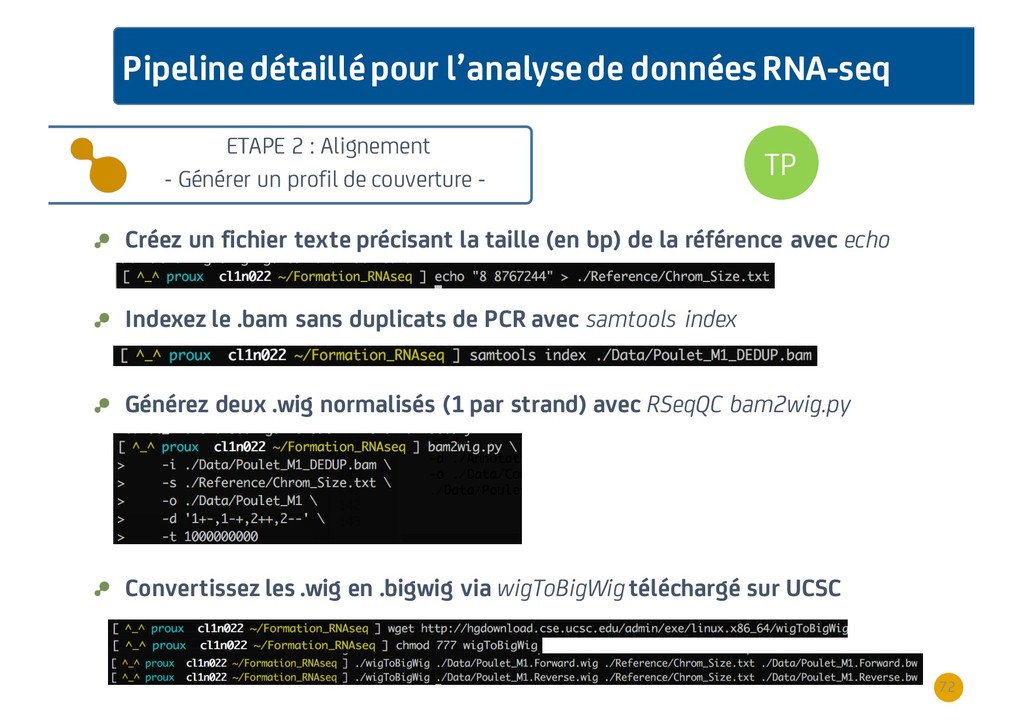{kind=link}
{kind=link}
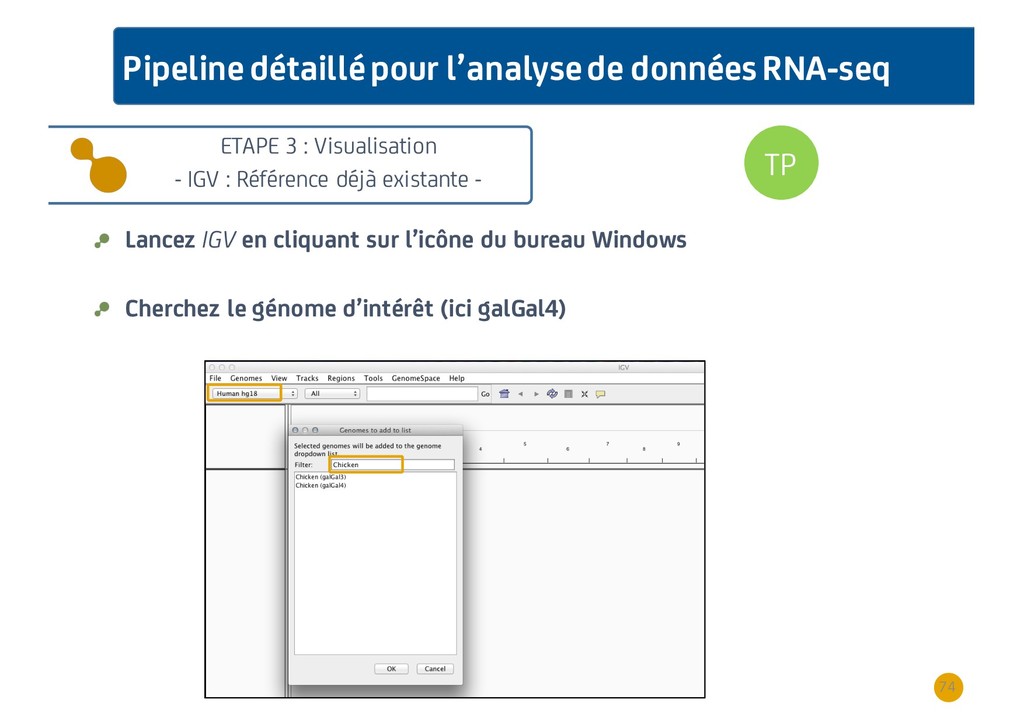{kind=link}
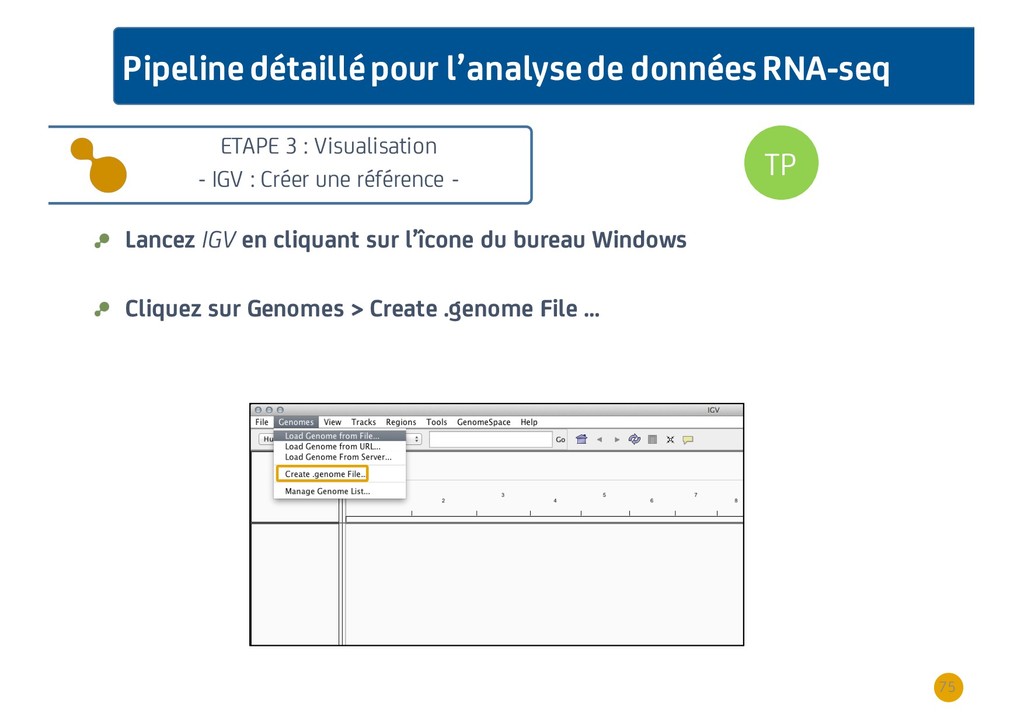{kind=link}
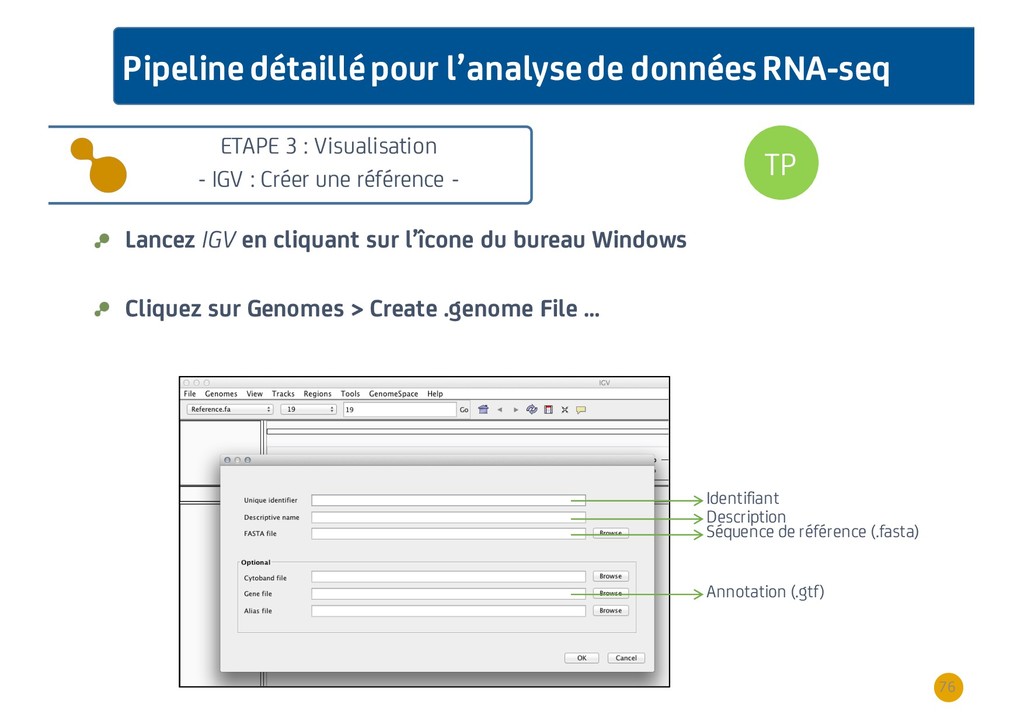{kind=link}
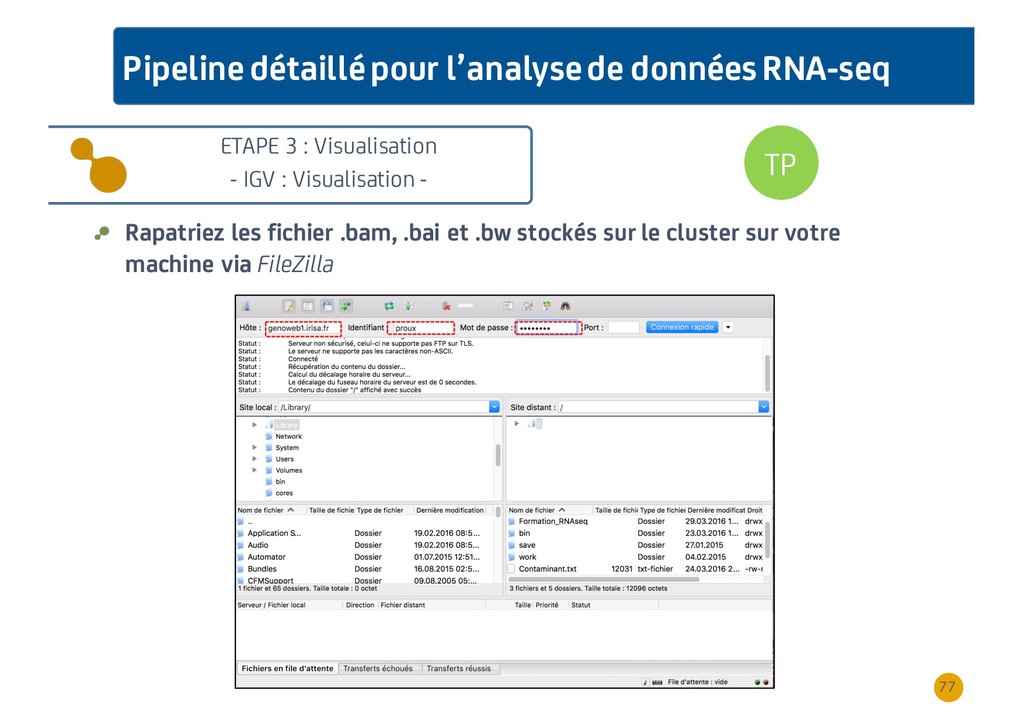{kind=link}
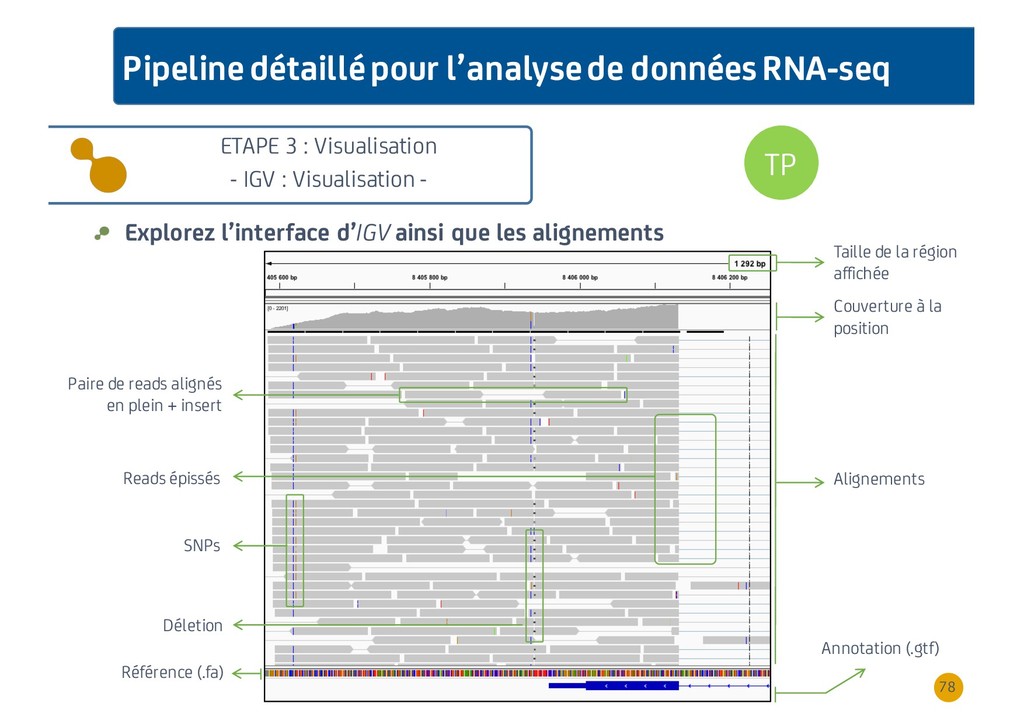{kind=link}
{kind=link}
{kind=link}
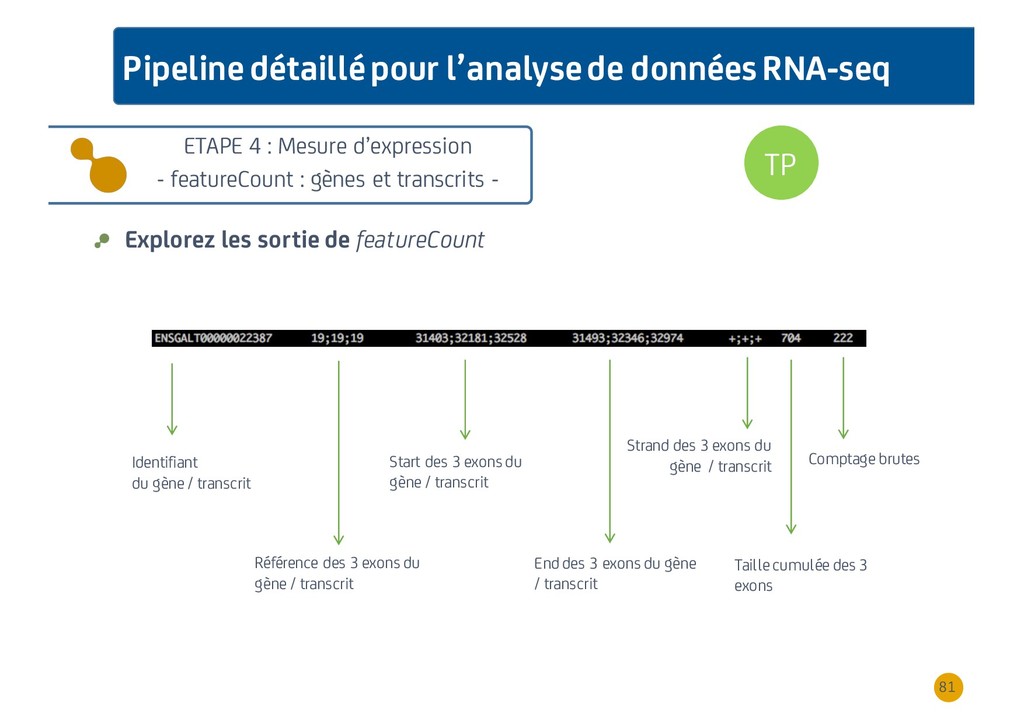{kind=link}
{kind=link}