- Analyse de données de ChIP-seq sous R 50 Qu’est ce que les formats WIG / BIGWIG / BEGRAPH ? • Format utiliser pour stocker des informations de visualisation denses et continues (e.g. contenu en GC, probabilité, couverture en reads) • BIGWIG : format binaire, plus léger et en général recommandé • WIG : format ASCII, données continues • BEDGRAPH : format ASCII, données sparses Qu’est ce que le format BED ? • « Browser Extensible Data » • Format tabulaire adapté à l’affichage de données d’intervalles sur les « genome browsers » (UCSC, Ensembl, IGV …)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
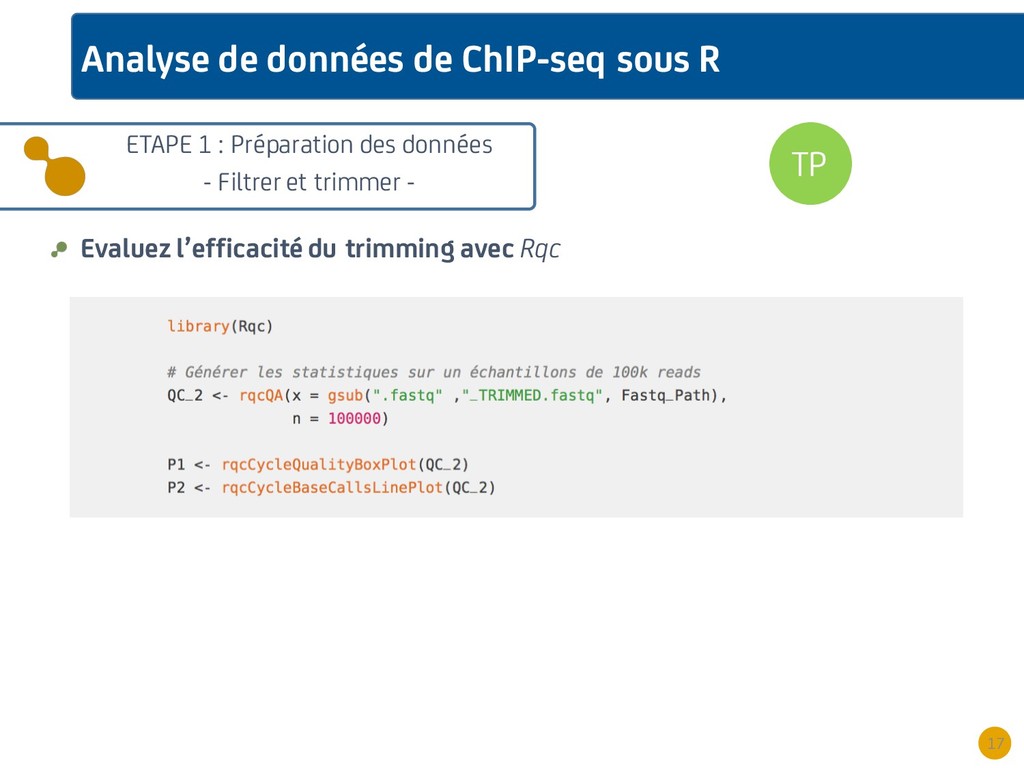{kind=link}
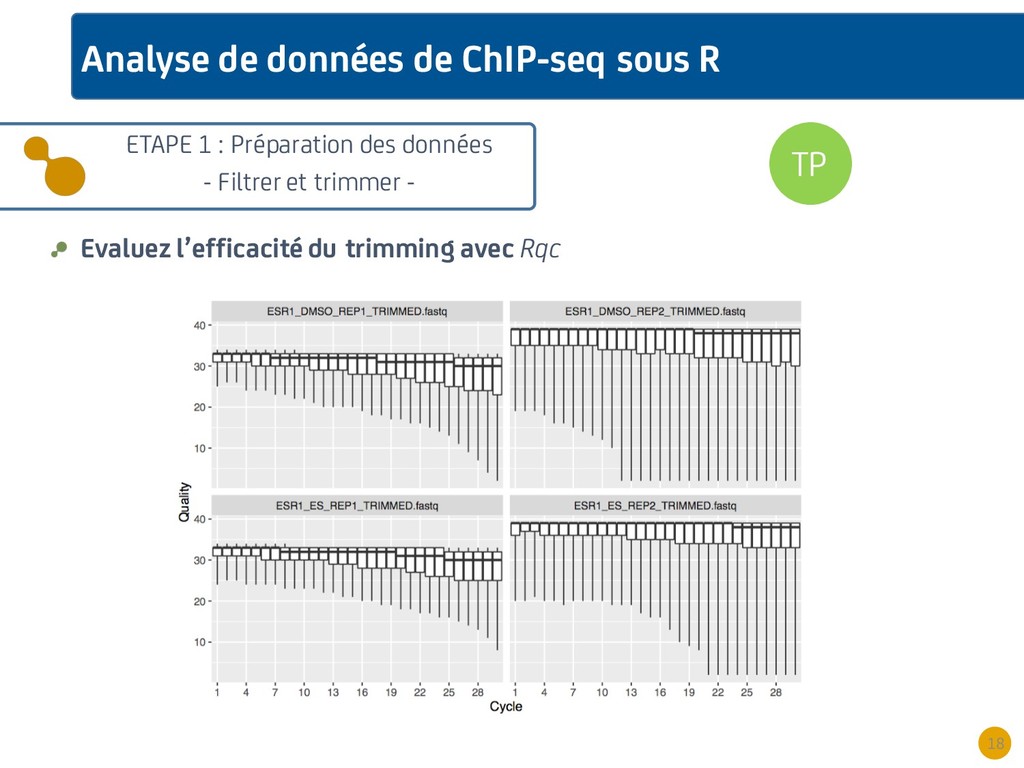{kind=link}
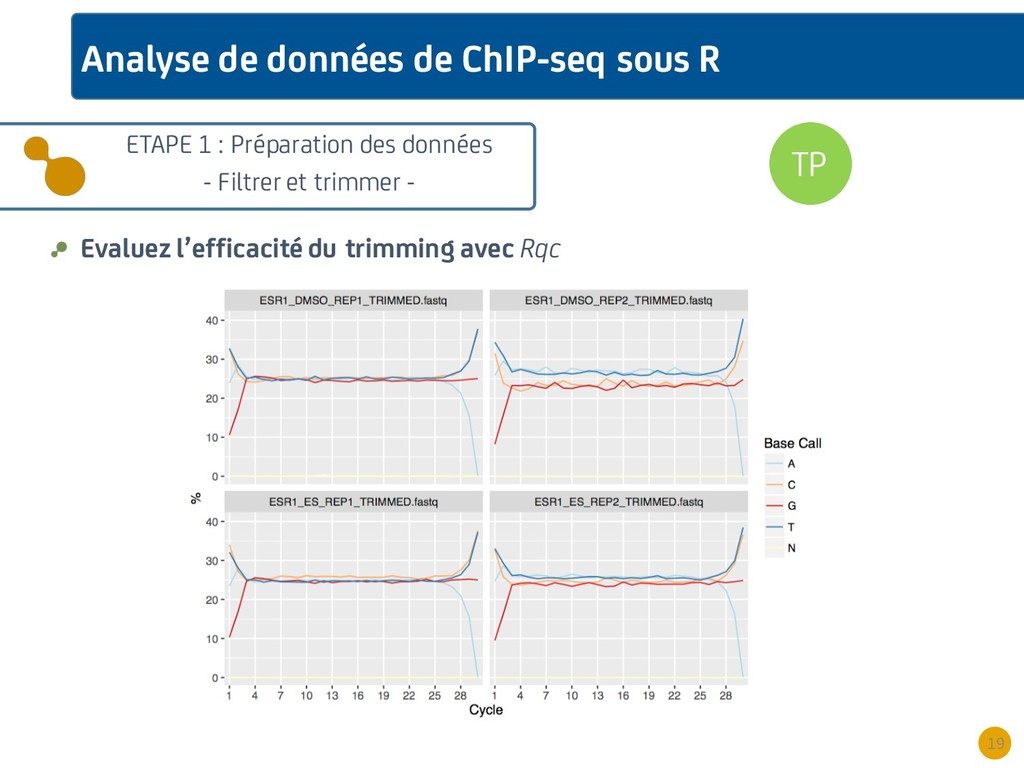{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
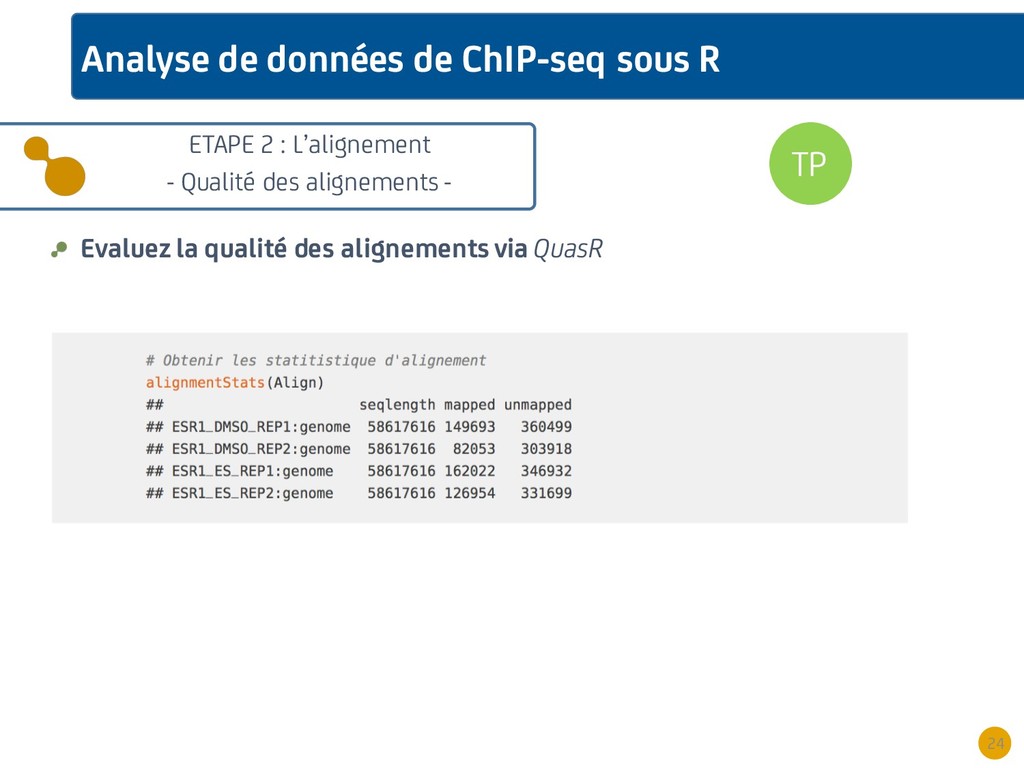{kind=link}
{kind=link}
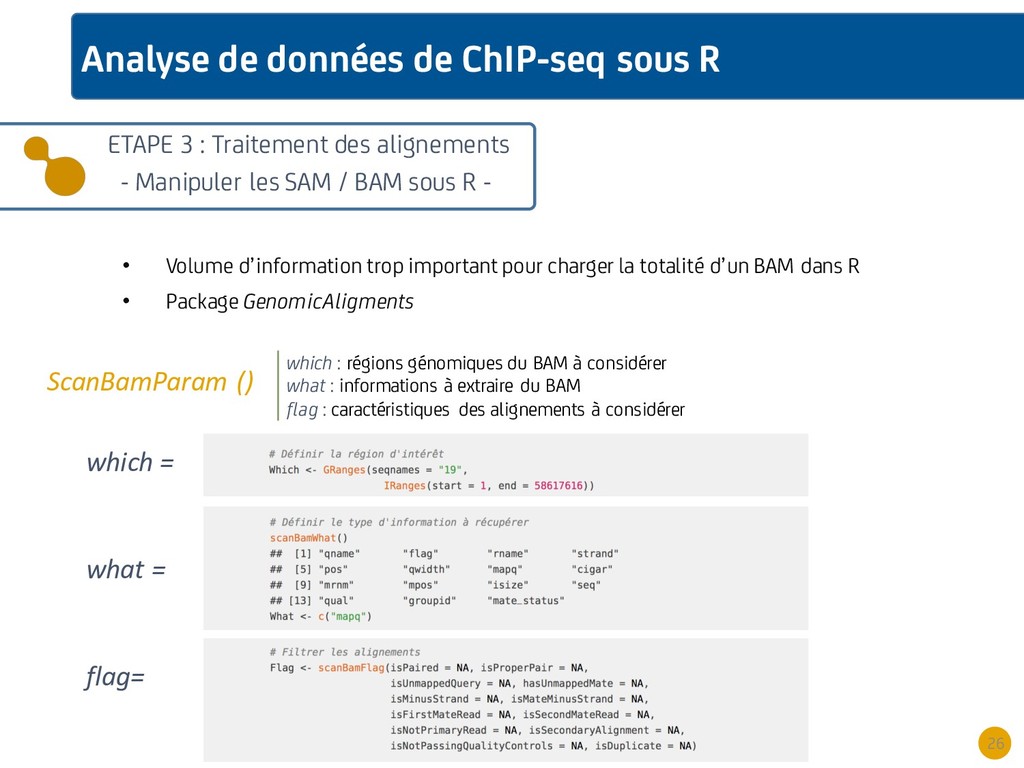{kind=link}
{kind=link}
{kind=link}
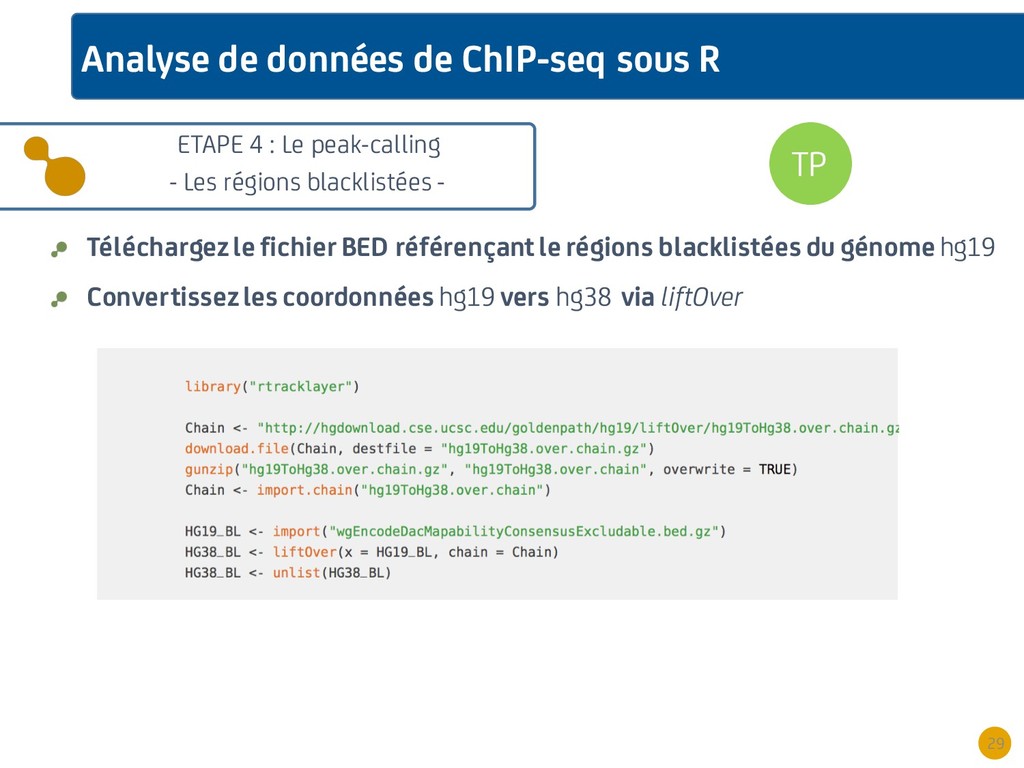{kind=link}
{kind=link}
{kind=link}
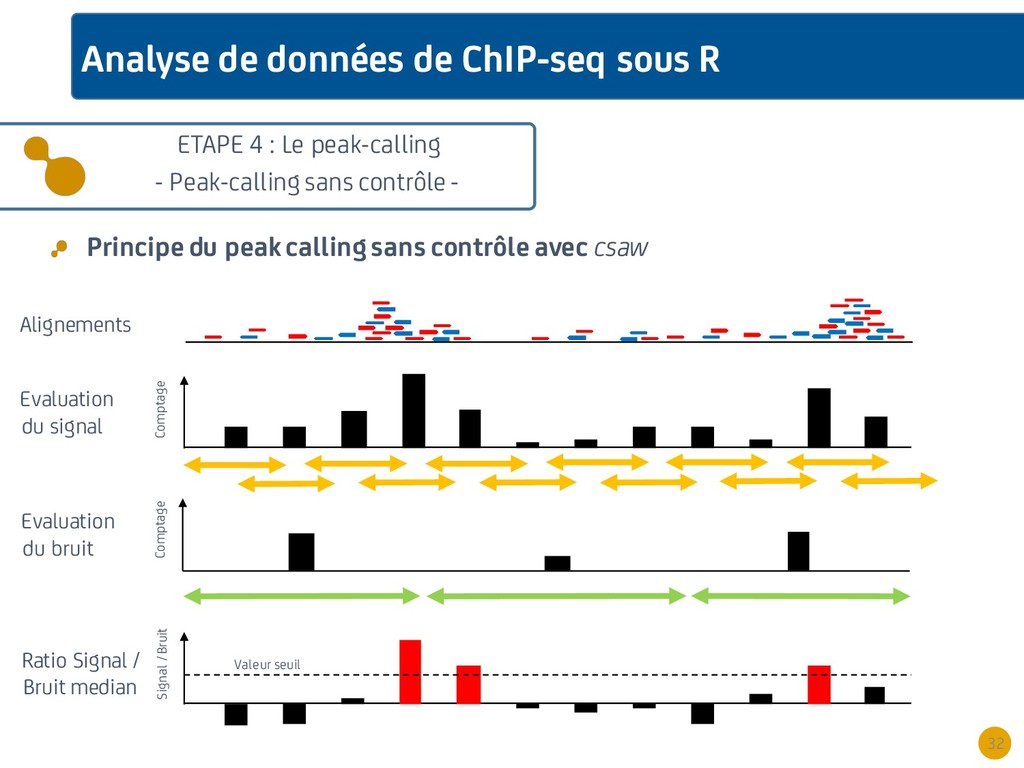{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
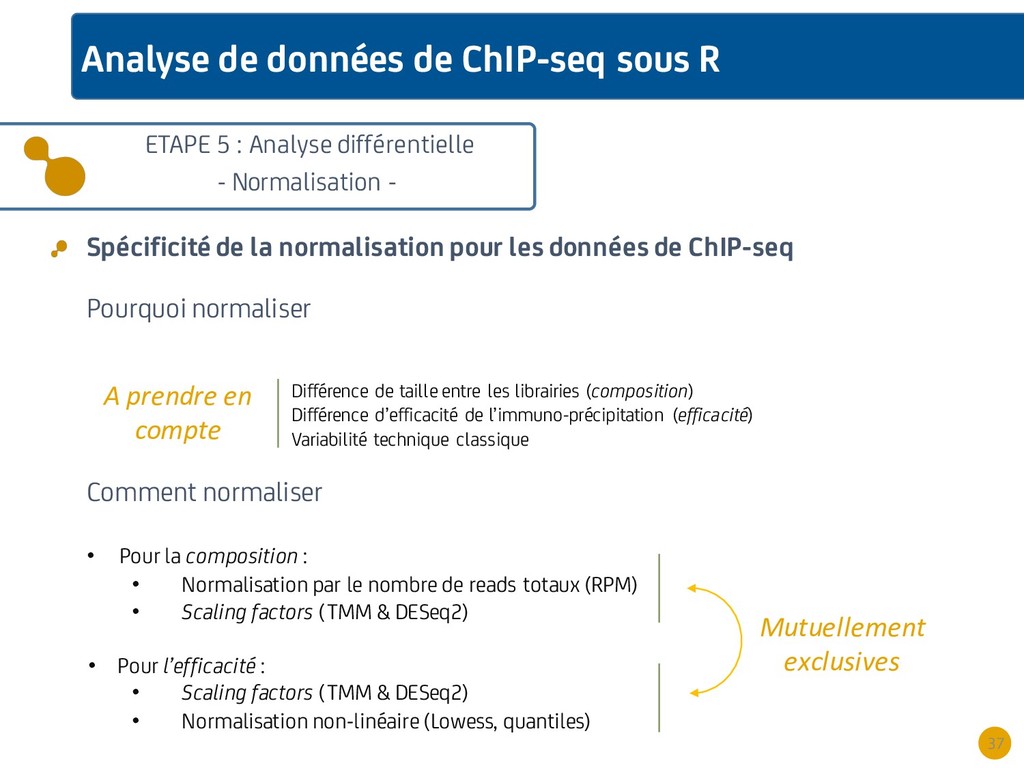{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
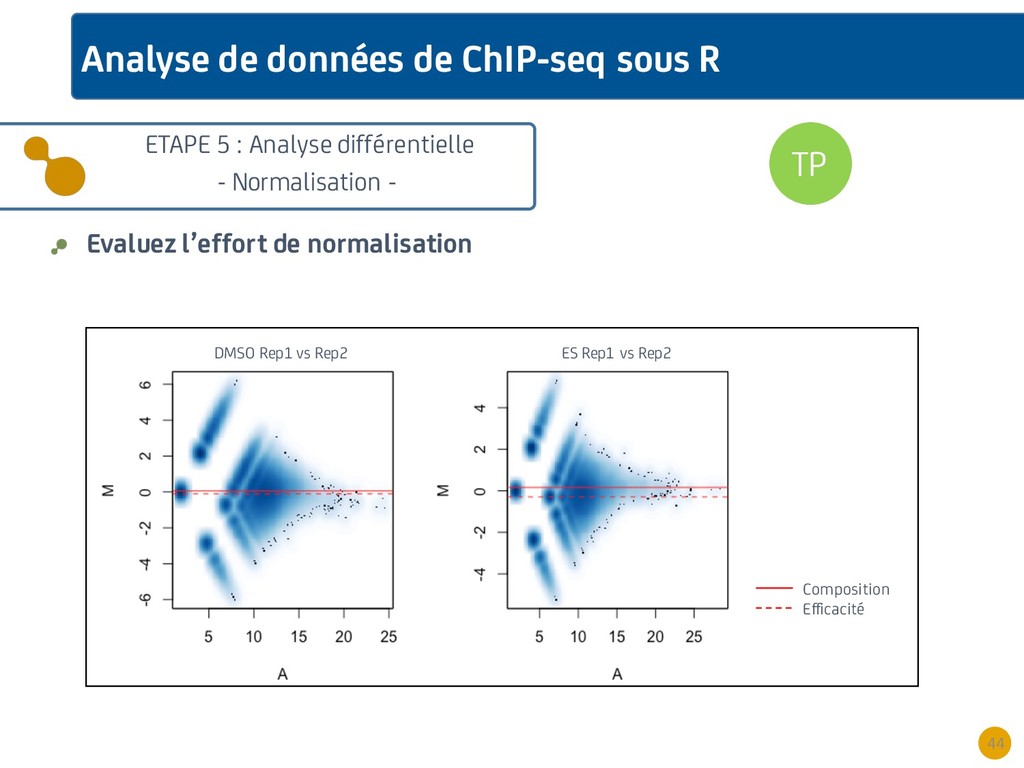{kind=link}
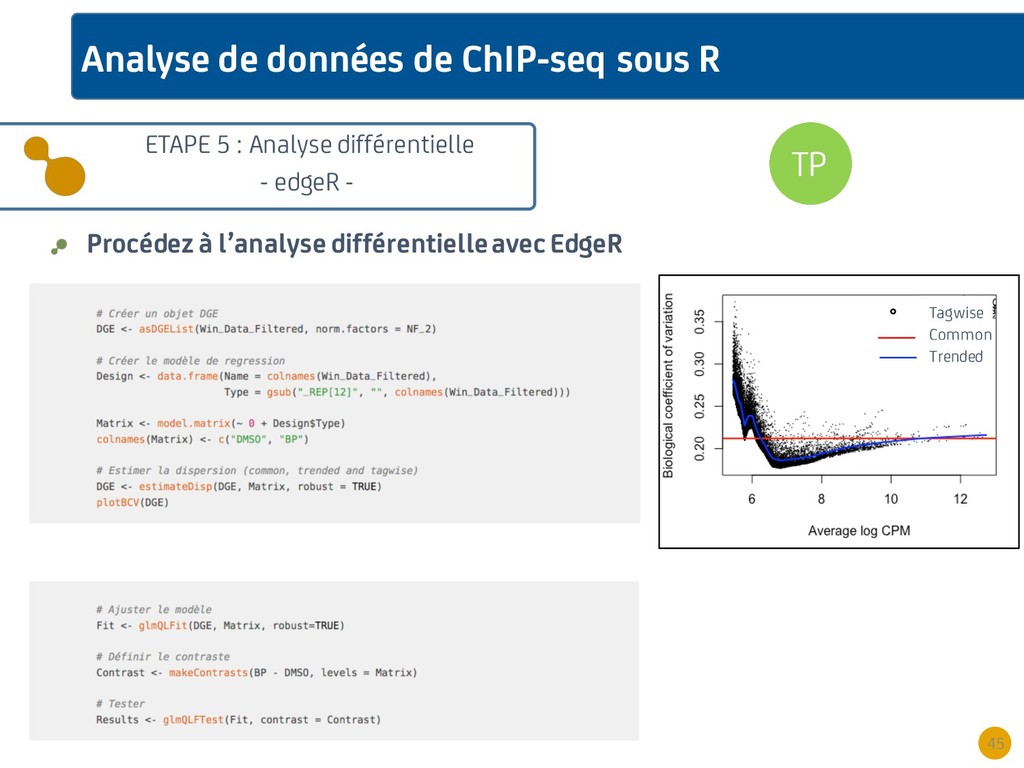{kind=link}
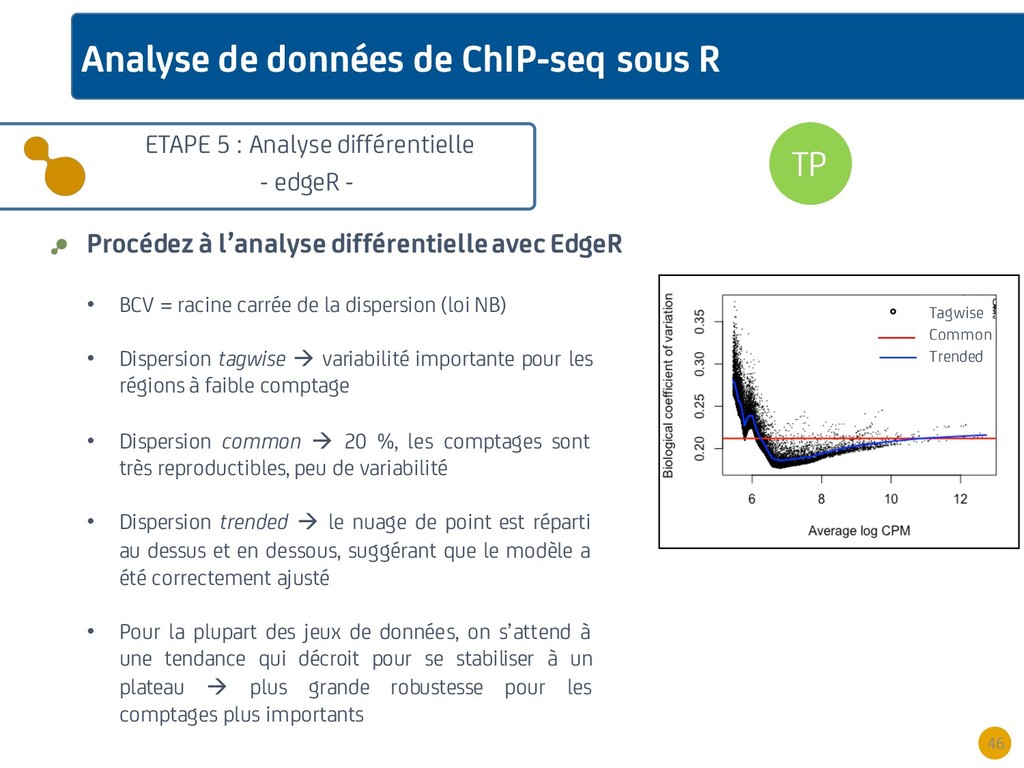{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
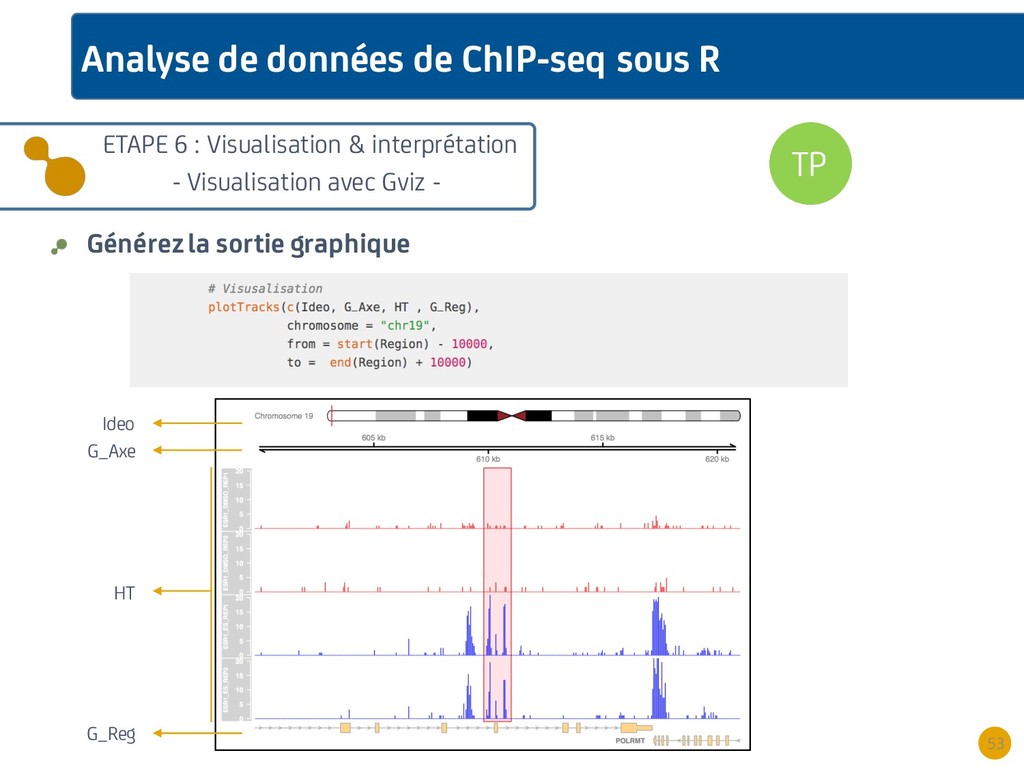{kind=link}
{kind=link}
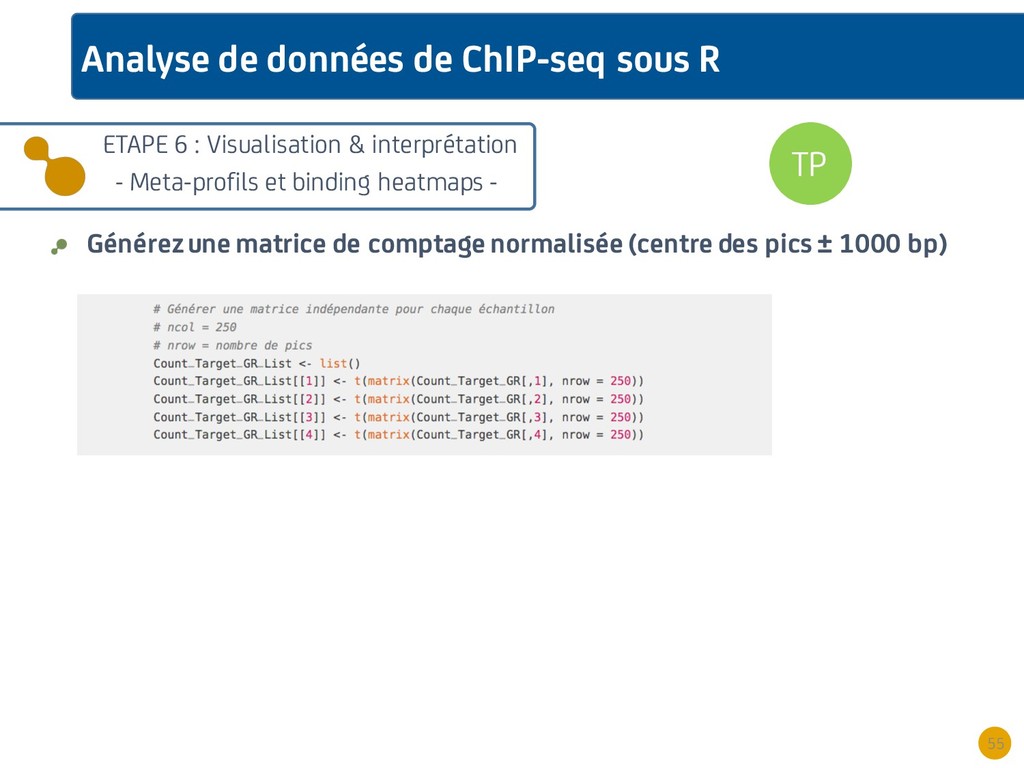{kind=link}
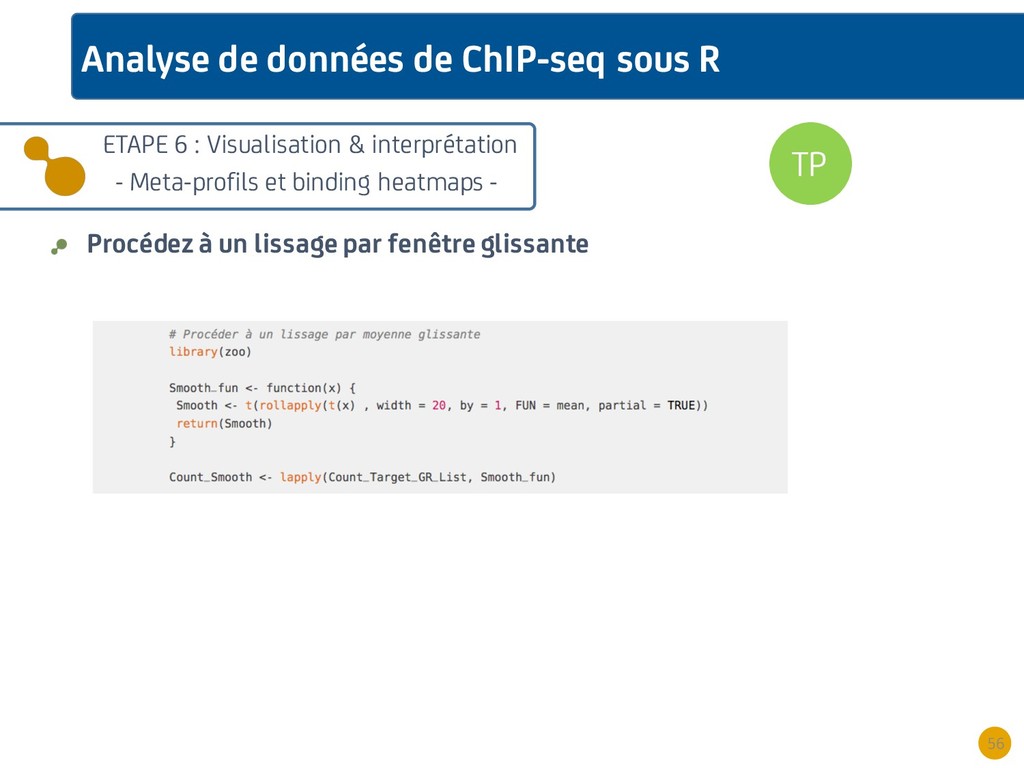{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
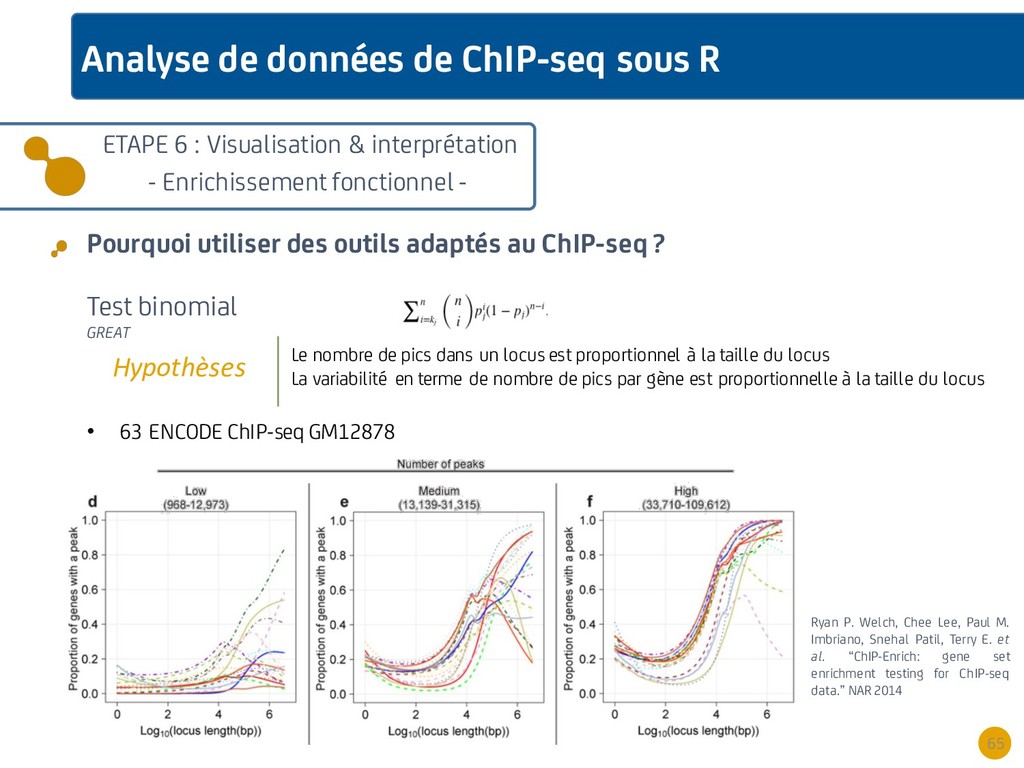{kind=link}
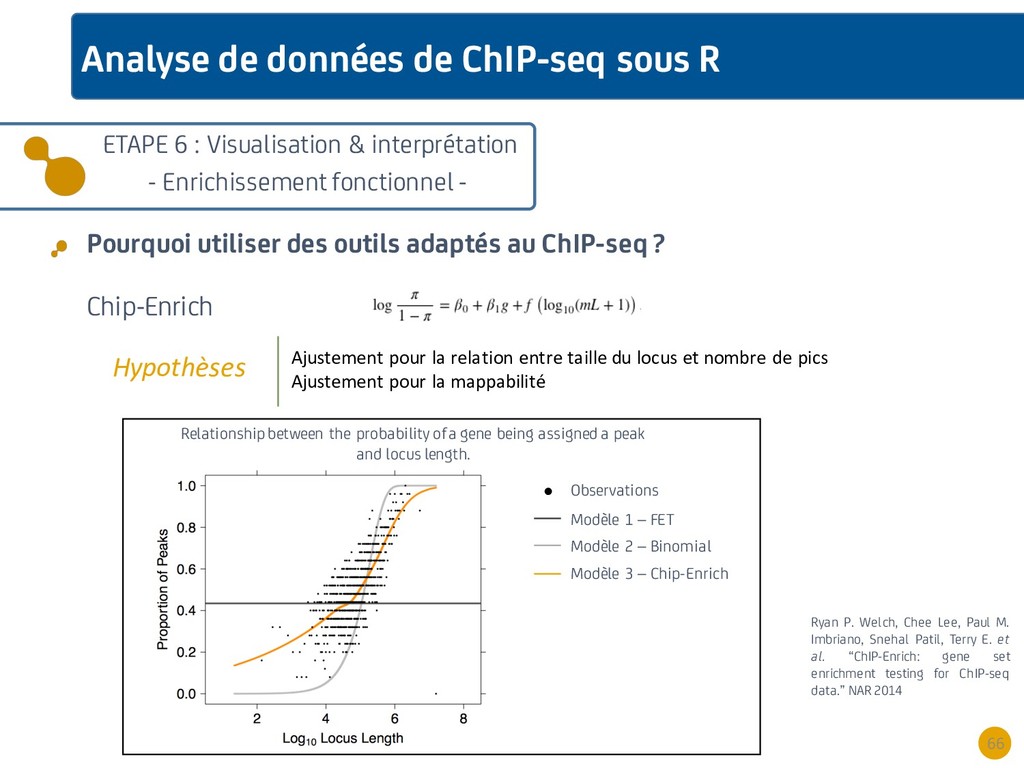{kind=link}
{kind=link}
{kind=link}