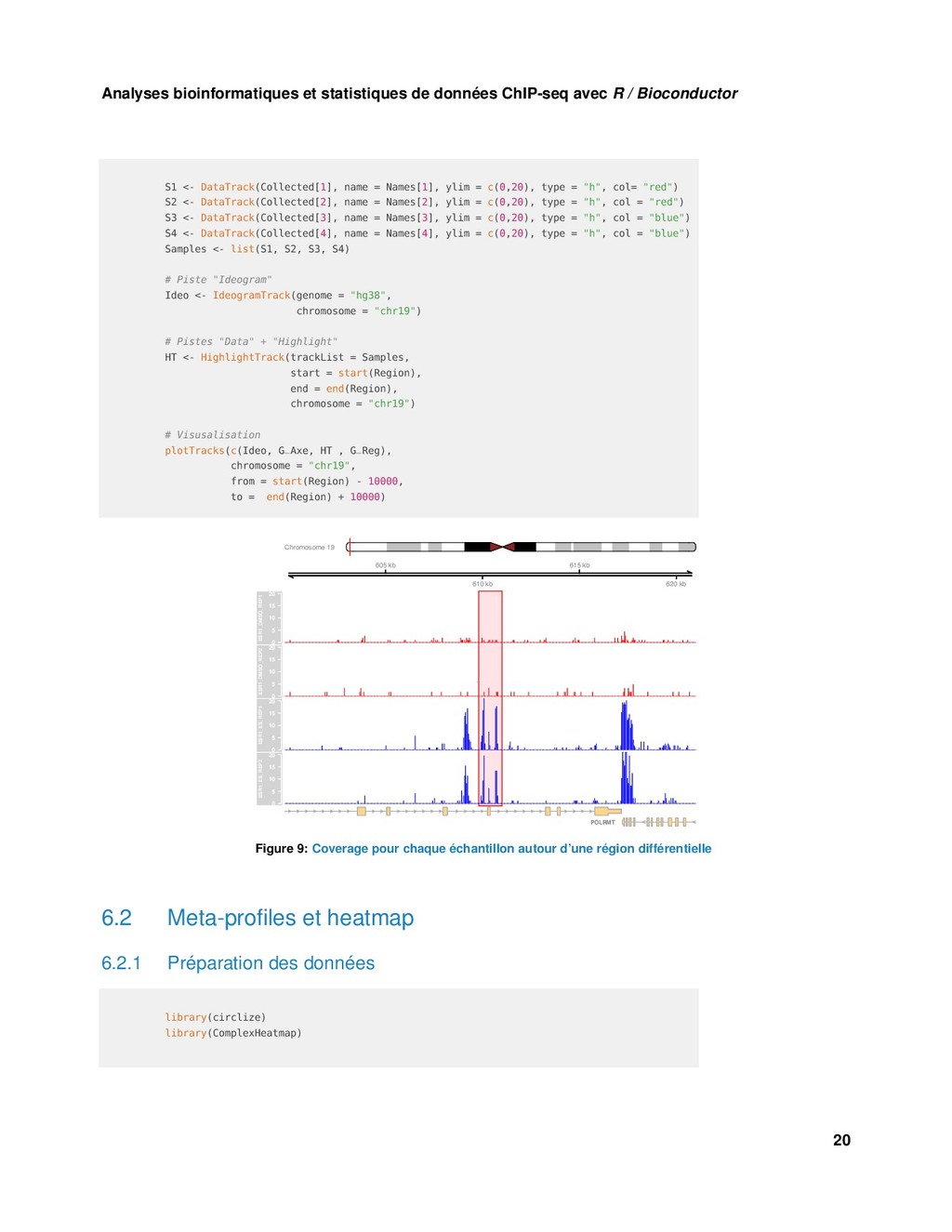
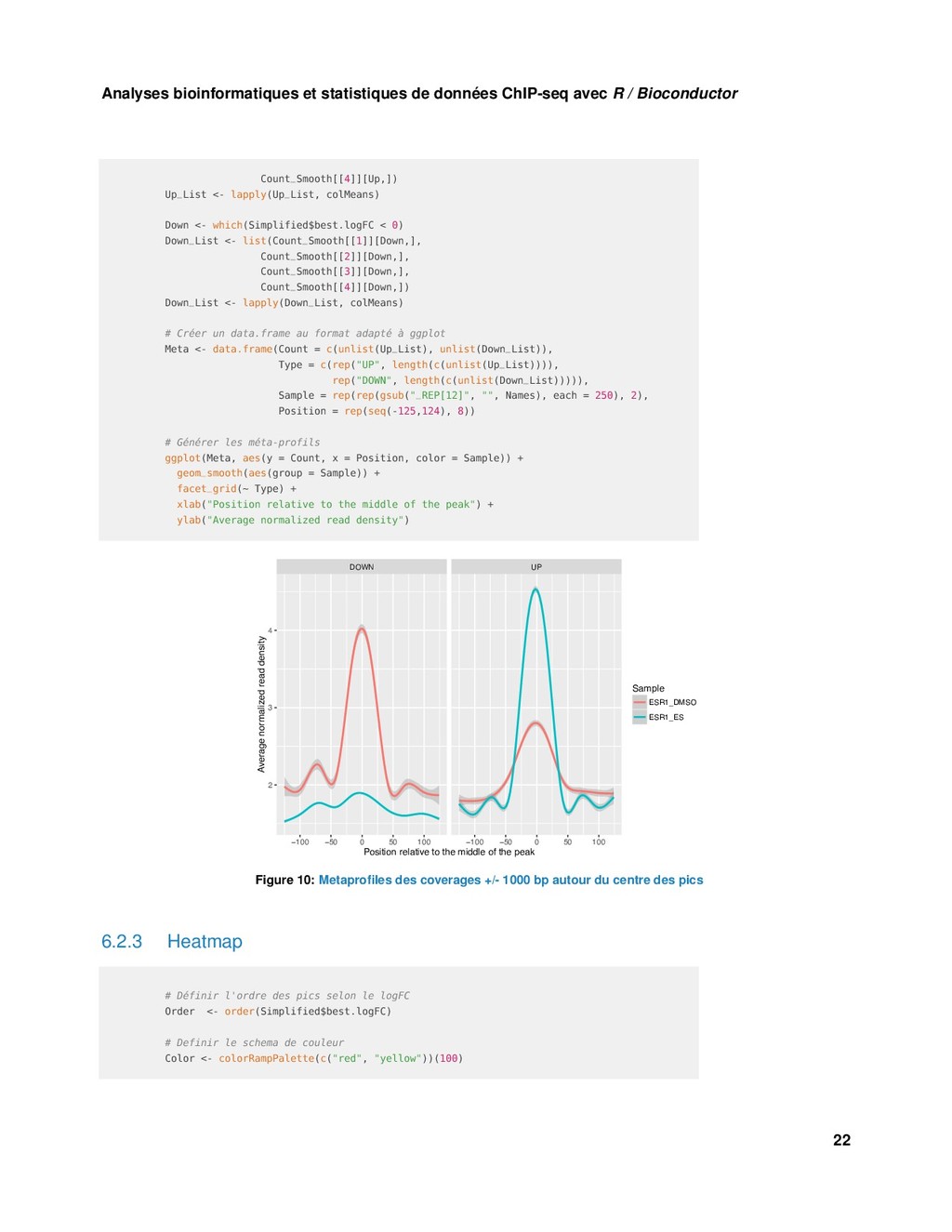
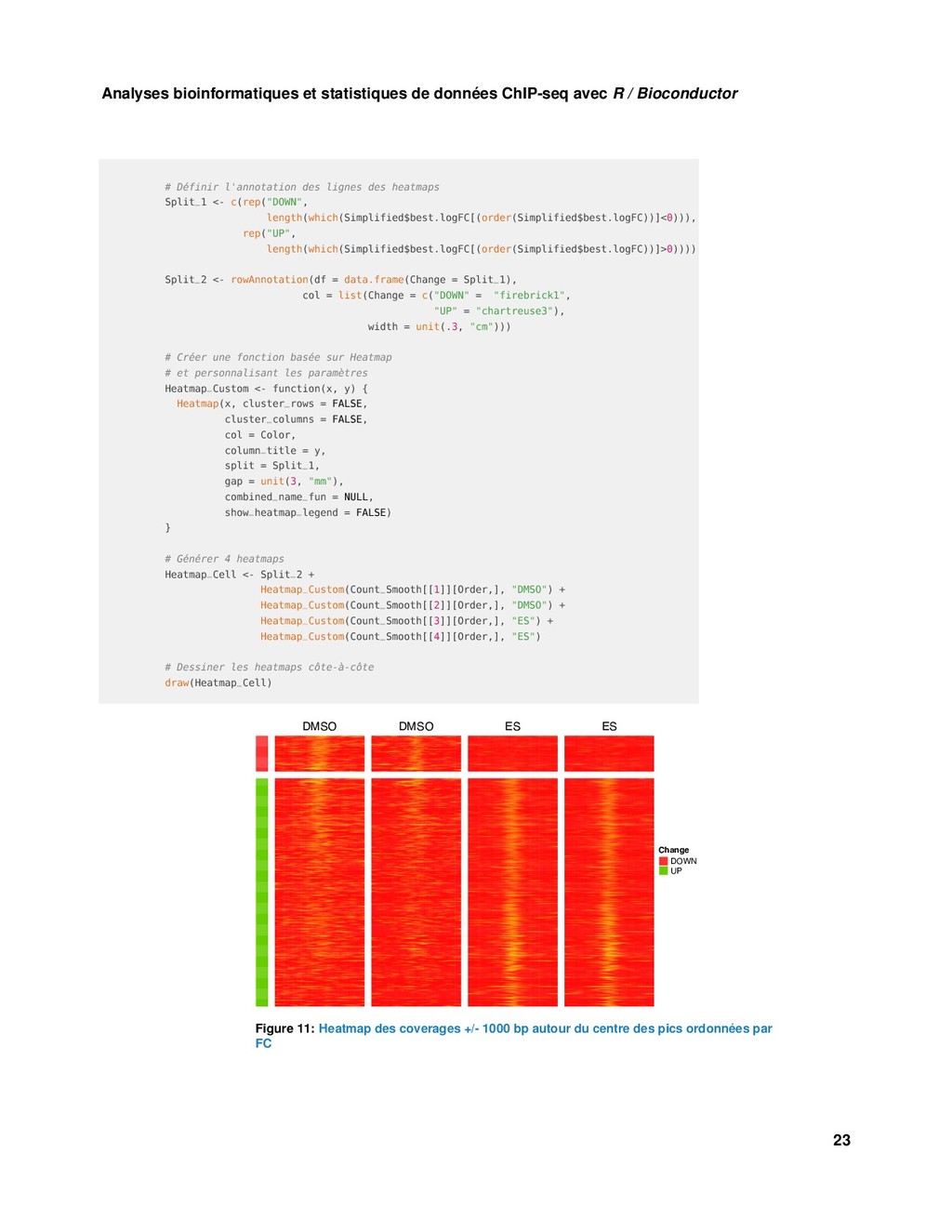
Bioconductor S1 <- DataTrack(Collected[1], name = Names[1], ylim = c(0,20), type = "h", col= "red") S2 <- DataTrack(Collected[2], name = Names[2], ylim = c(0,20), type = "h", col = "red") S3 <- DataTrack(Collected[3], name = Names[3], ylim = c(0,20), type = "h", col = "blue") S4 <- DataTrack(Collected[4], name = Names[4], ylim = c(0,20), type = "h", col = "blue") Samples <- list(S1, S2, S3, S4) # Piste "Ideogram" Ideo <- IdeogramTrack(genome = "hg38", chromosome = "chr19") # Pistes "Data" + "Highlight" HT <- HighlightTrack(trackList = Samples, start = start(Region), end = end(Region), chromosome = "chr19") # Visusalisation plotTracks(c(Ideo, G_Axe, HT , G_Reg), chromosome = "chr19", from = start(Region) - 10000, to = end(Region) + 10000) Chromosome 19 605 kb 610 kb 615 kb 620 kb 0 5 10 15 20 ESR1_DMSO_REP1 0 5 10 15 20 ESR1_DMSO_REP2 0 5 10 15 20 ESR1_ES_REP1 0 5 10 15 20 ESR1_ES_REP2 POLRMT Figure 9: Coverage pour chaque échantillon autour d’une région différentielle 6.2 Meta-profiles et heatmap 6.2.1 Préparation des données library(circlize) library(ComplexHeatmap) 20
{kind=link}
{kind=link}
{kind=link}
{kind=link}
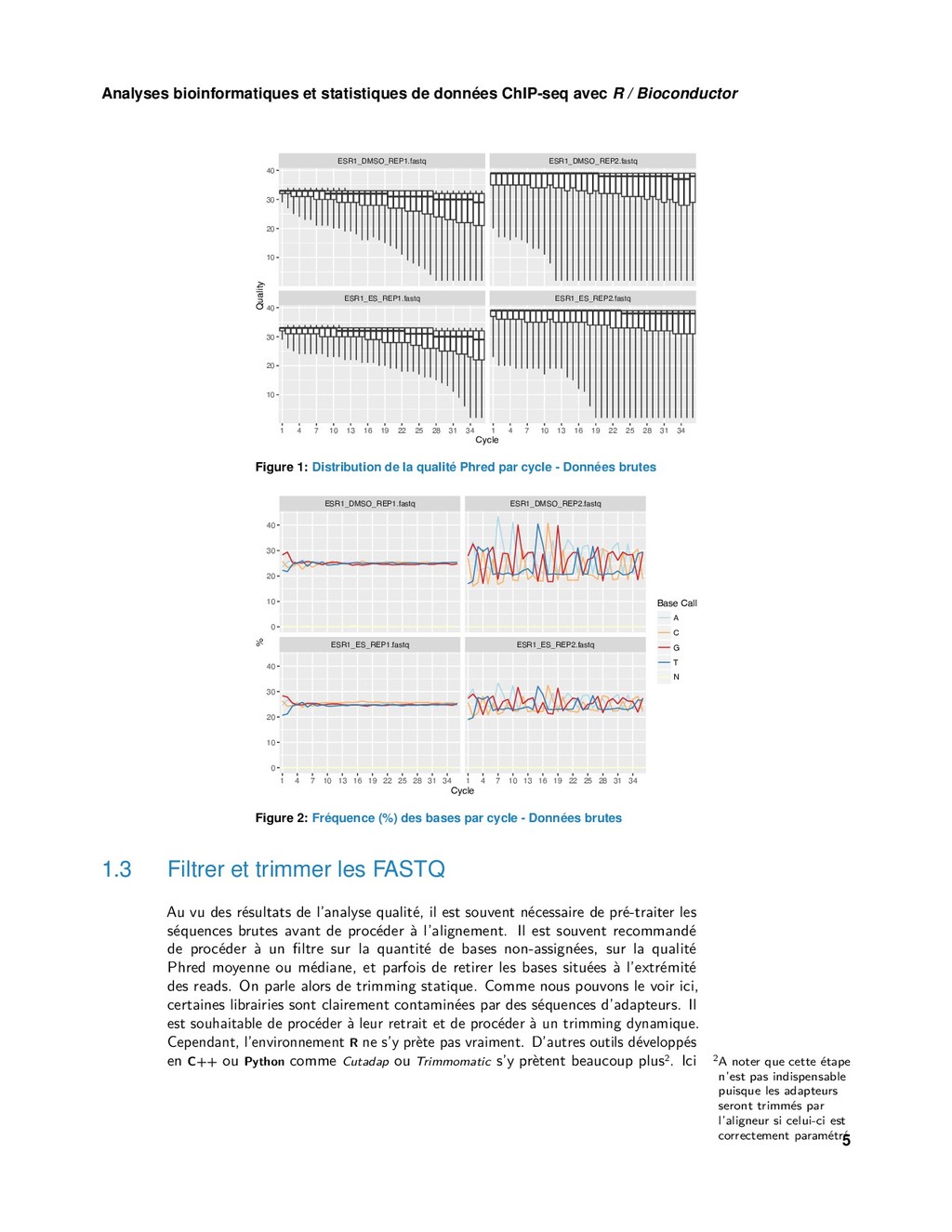{kind=link}
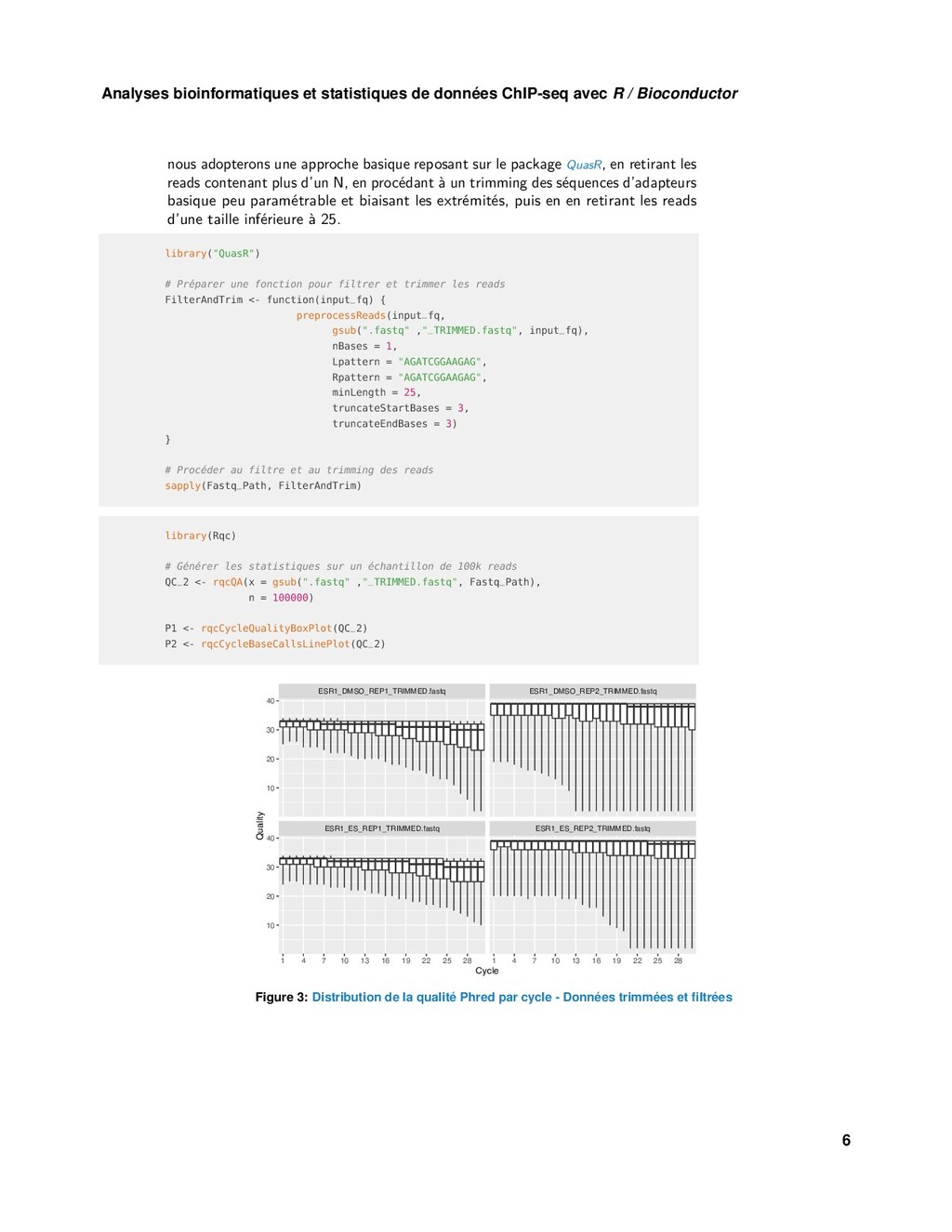{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
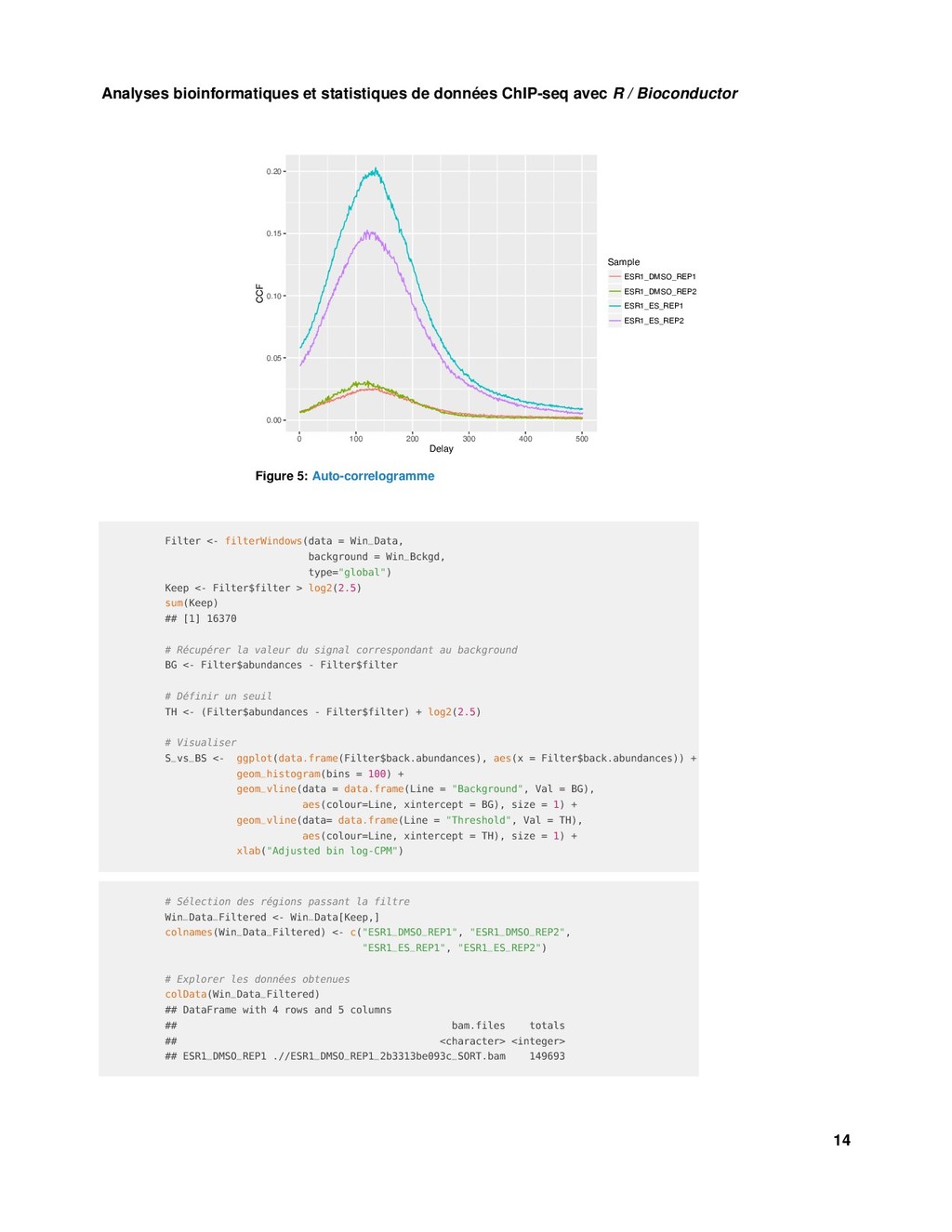{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}