sequences to look for the presence of RNA pol2 pausing pattern. • To identify and quantify the number of R-loops and Tandem repeats in samples of promoter sequences of 50 genes sampled randomly from each chromosome • To establish a correlation between the presence of R-loops and Tandem repeats with the instance of pausing
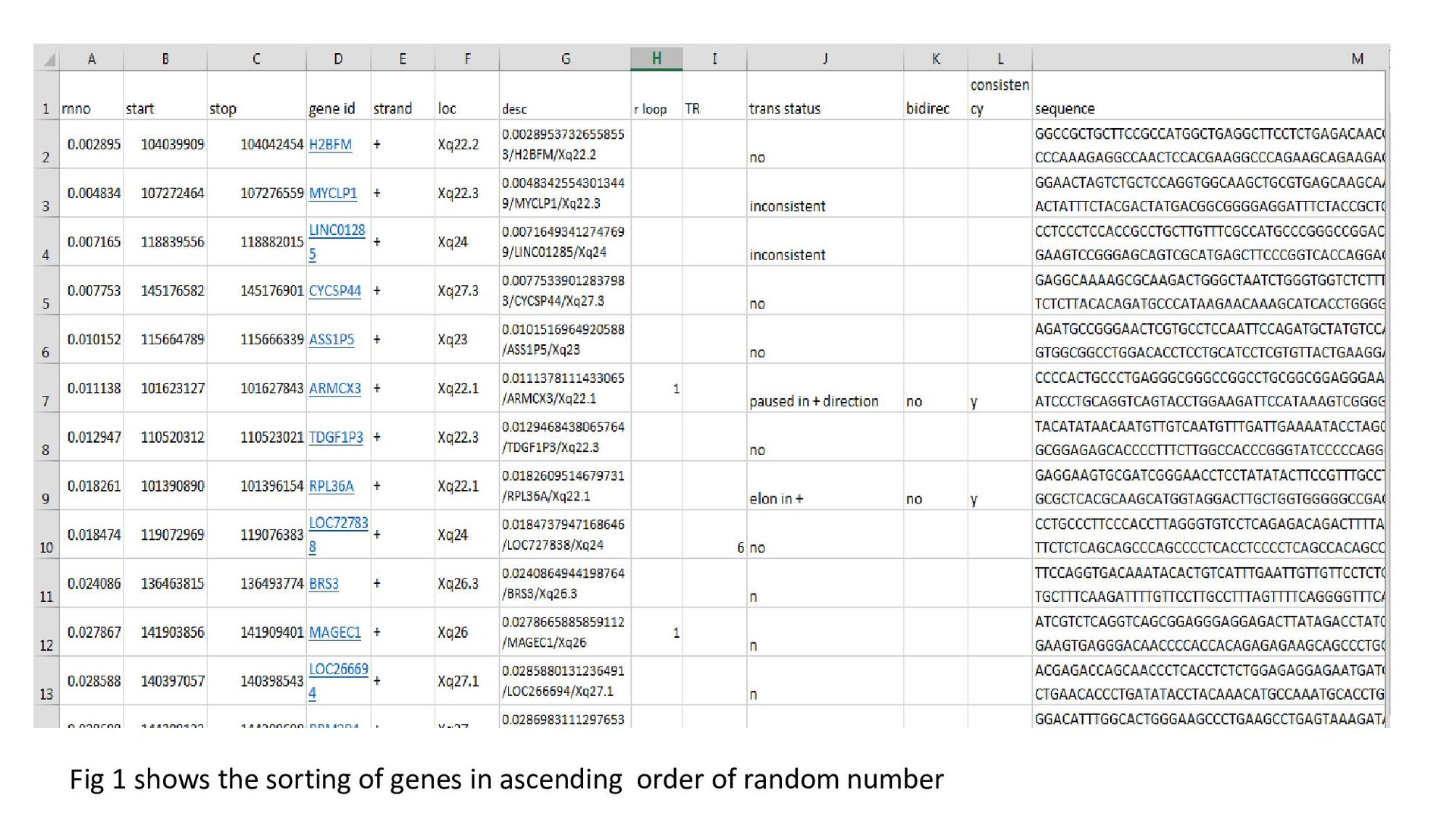
used: GRCh38.p2 annotation release 105 • The assembly was accessed using NCBI Map Viewer(Entrez Genome Viewer) • The gene list of each chromosome was collected and sorted according to the direction of transcription ( + / - orientation). The genes in + orientation were chosen for study • Using MS Excel functions, random numbers were assigned and the genes were sorted in ascending order of the random numbers.(fig.1) • First 50 genes were selected for study. First 1000 nucleotides collected for each gene
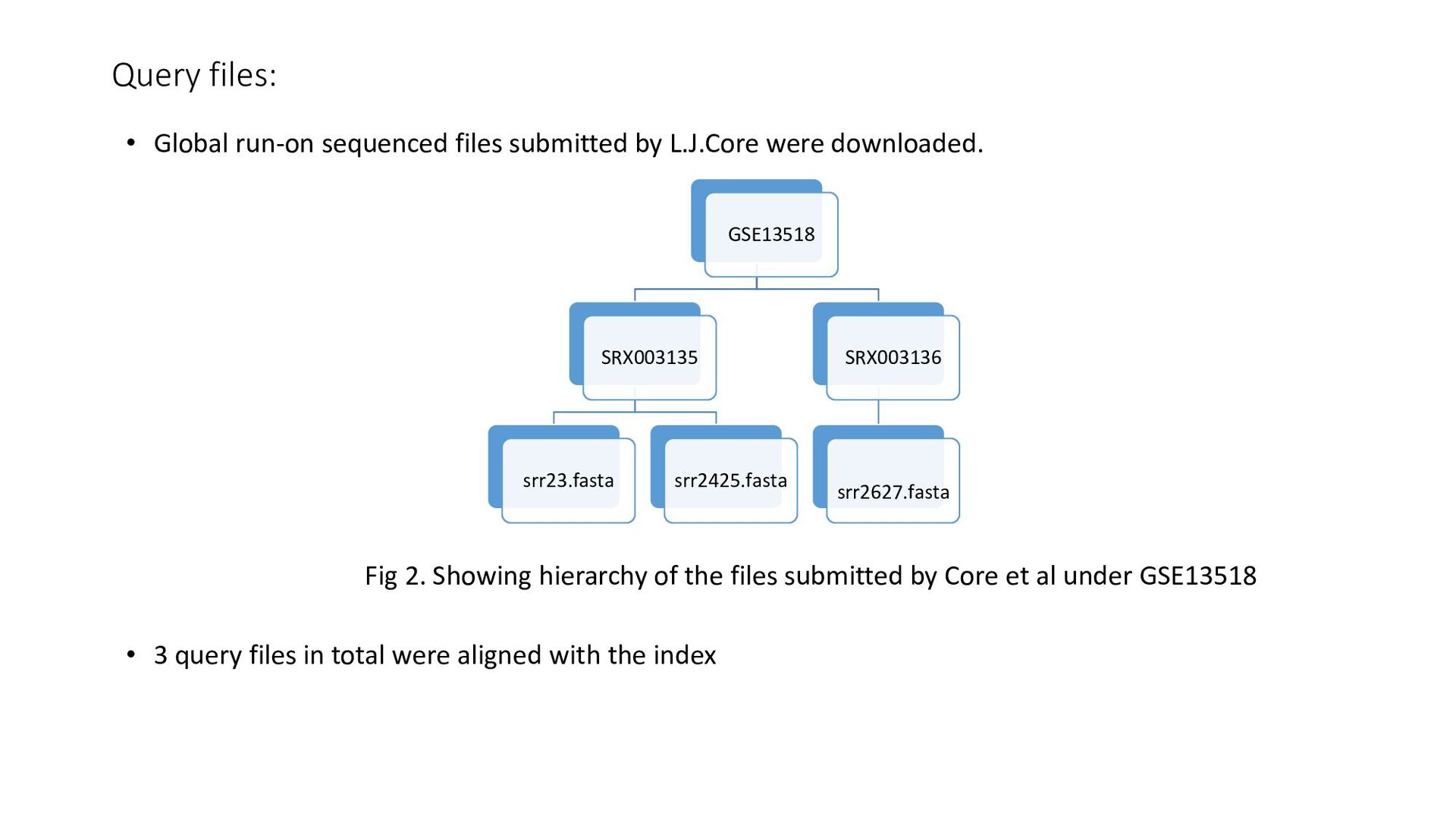
were downloaded. Fig 2. Showing hierarchy of the files submitted by Core et al under GSE13518 • 3 query files in total were aligned with the index GSE13518 SRX003135 srr23.fasta srr2425.fasta SRX003136 srr2627.fasta
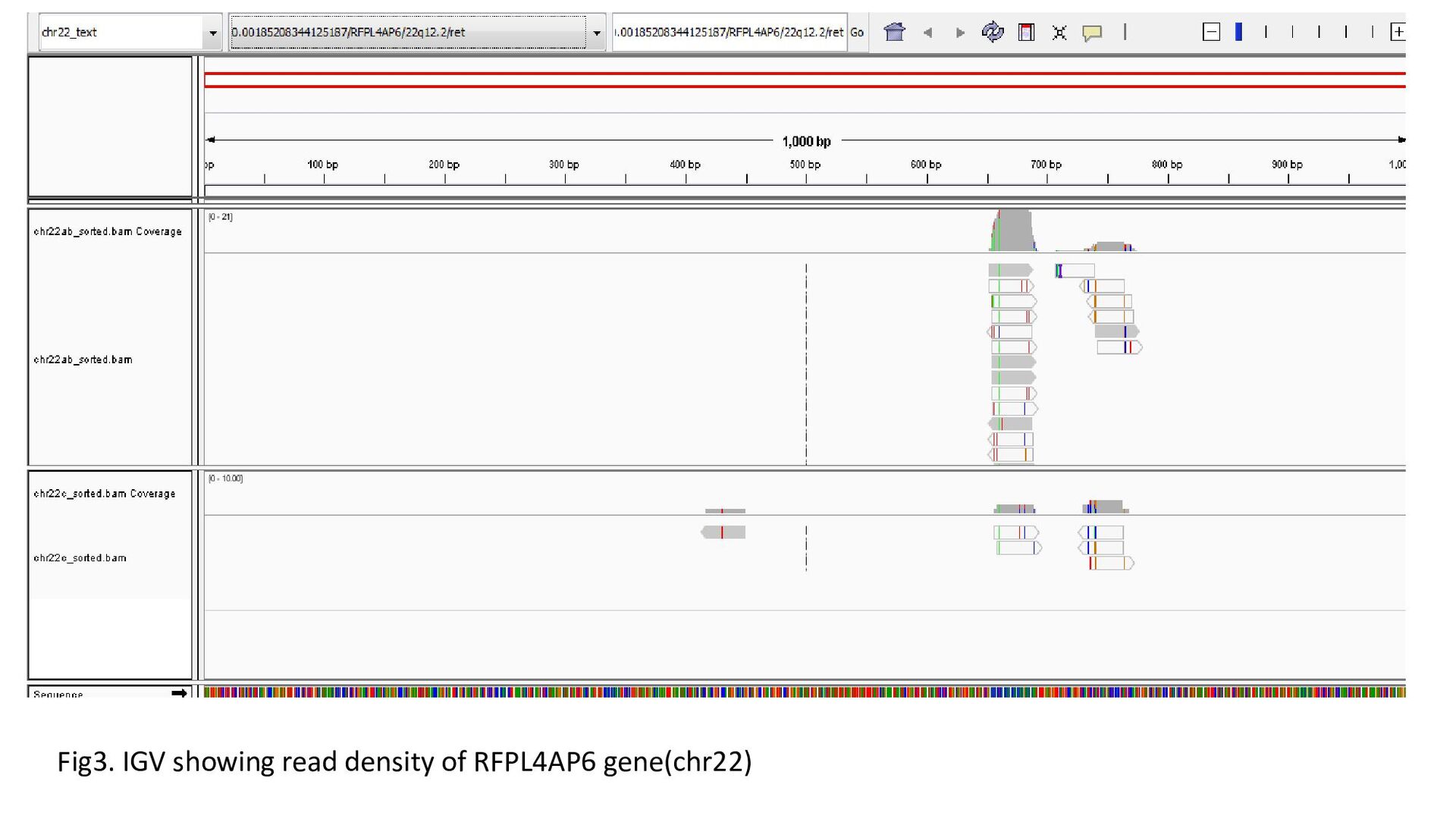
each chromosome were built as index files. They were named as chr1,2..Y. • Each of the 3 query files were aligned with index files. Alignment results were obtained in sam format. • 3 sam files obtained for each chromosome. Quantification of read density using Integrative Genomics Viewer • IGV supports bam format. Sam-> bam conversion was done using samtools. • Merging of the two query files(srr23.fasta and srr2425.fasta) was done to bring a single file representing the library srx003135 • Files were sorted and Index files created for each bam file
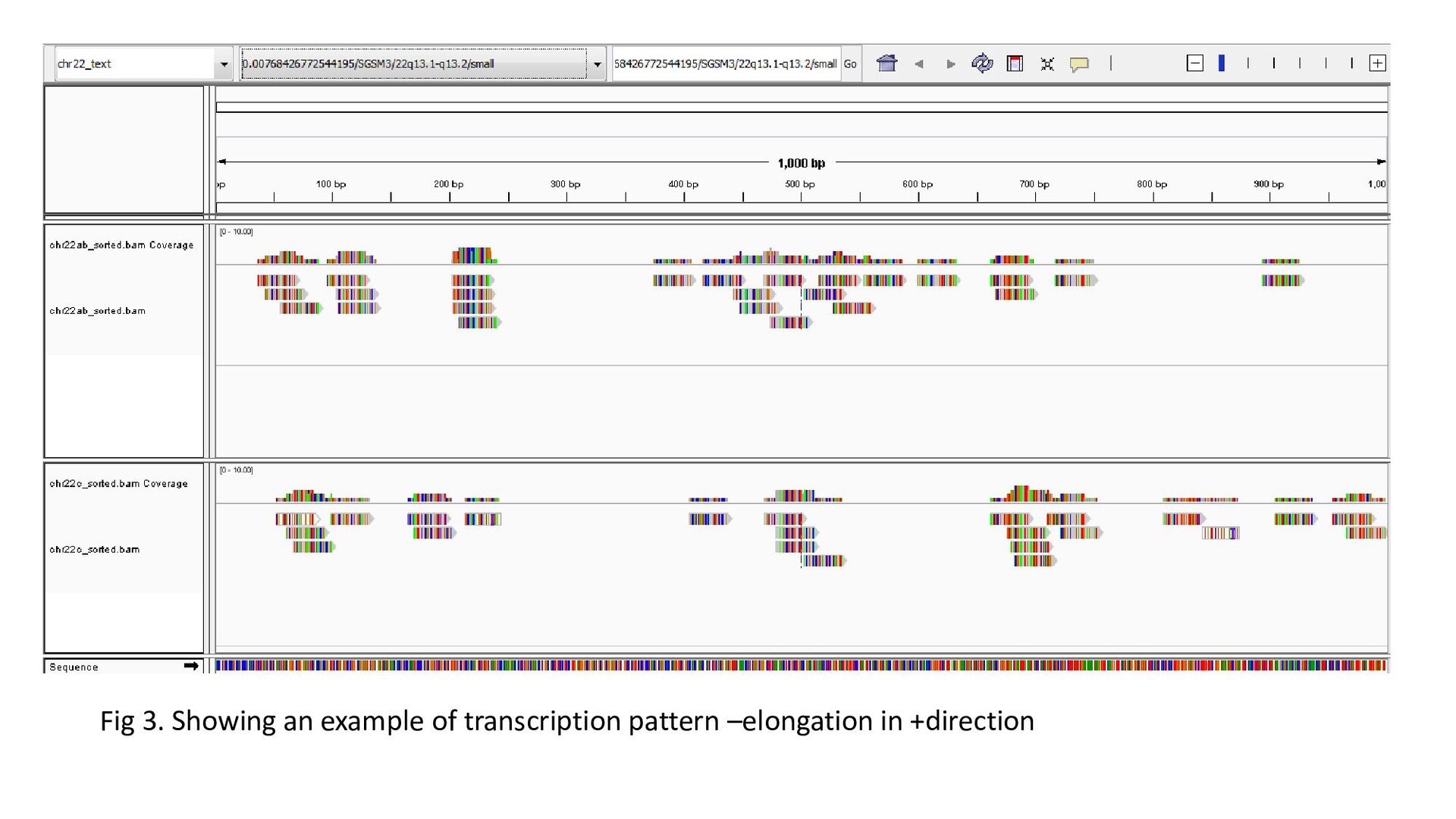
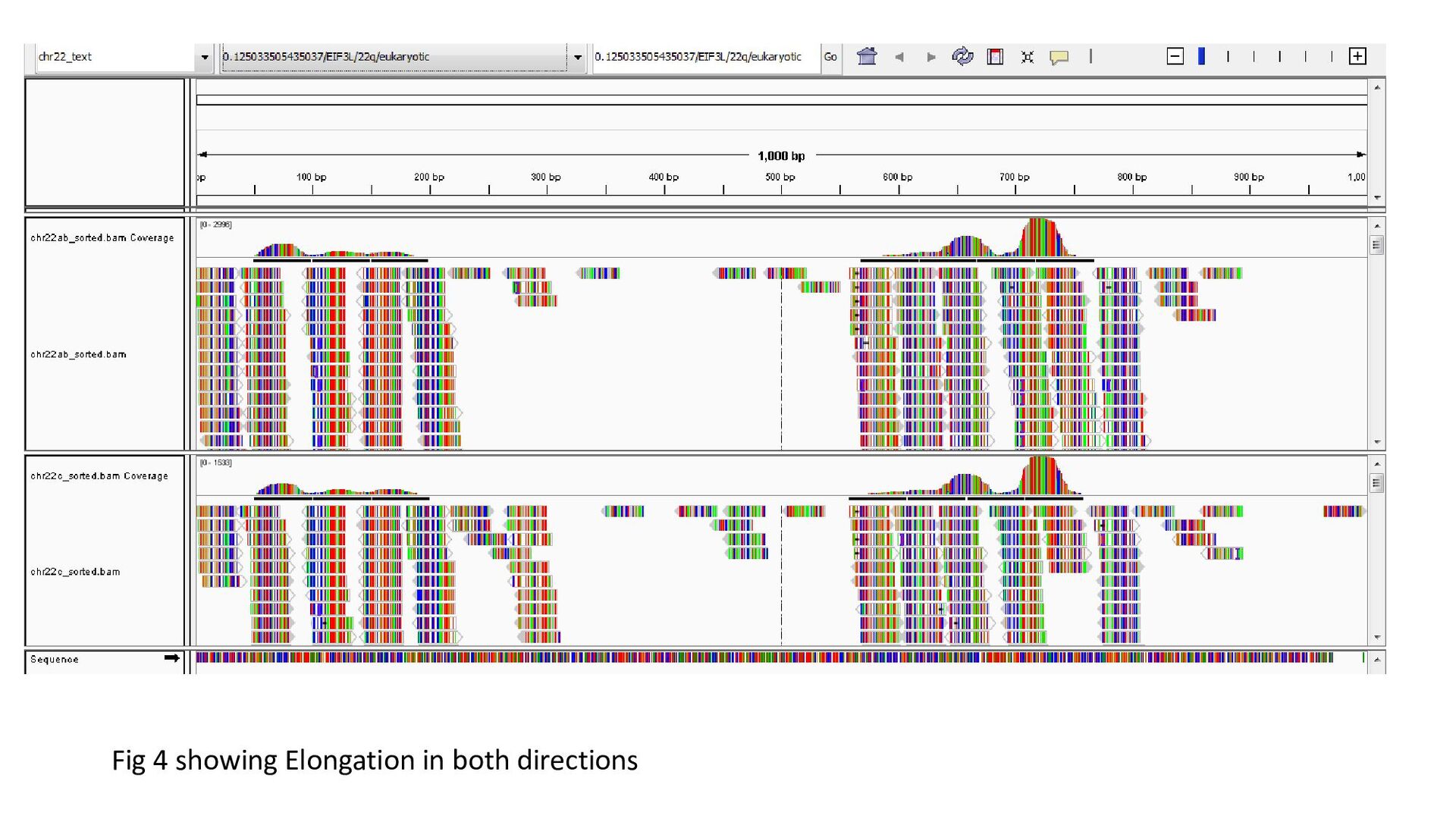
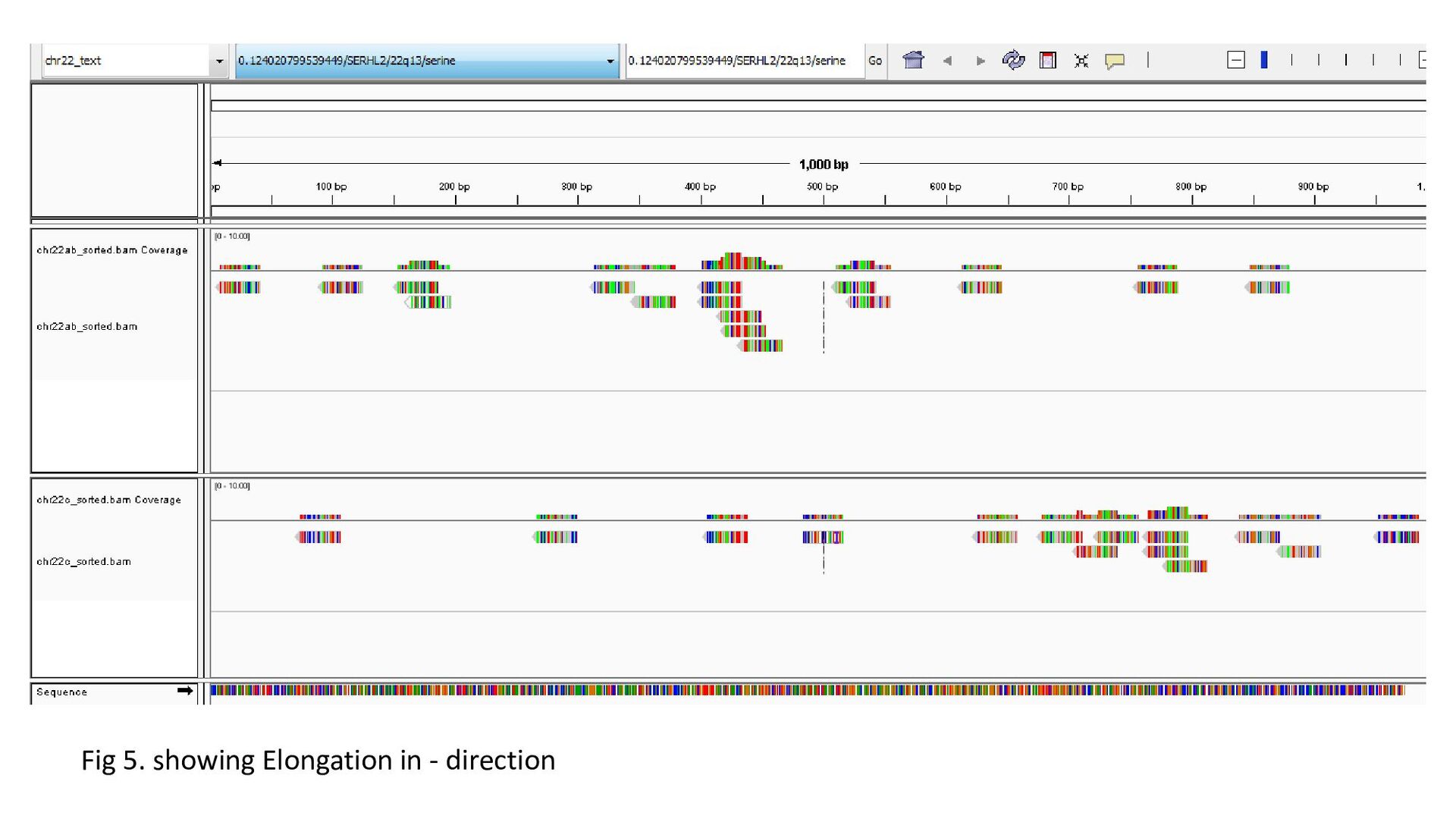
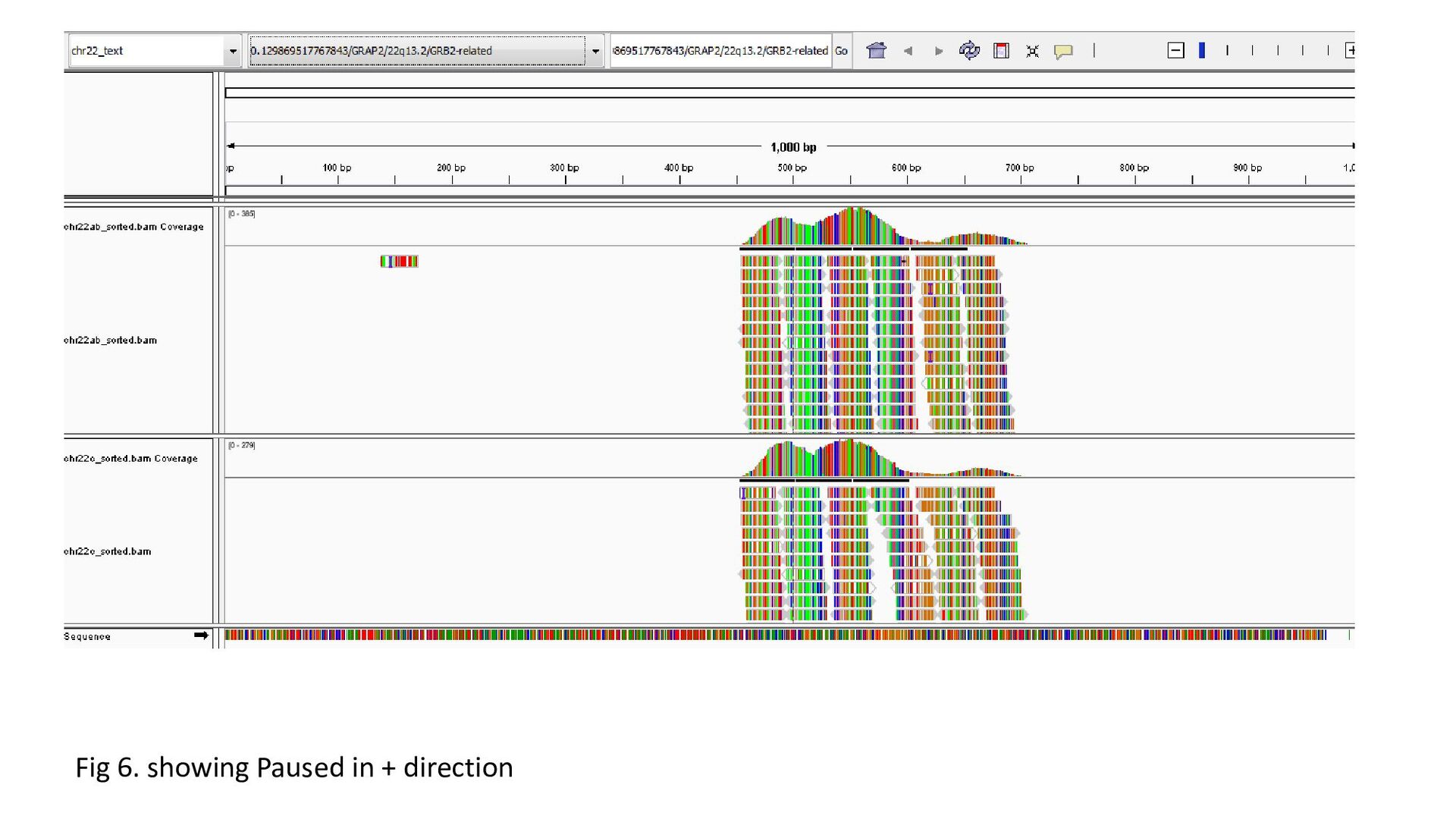
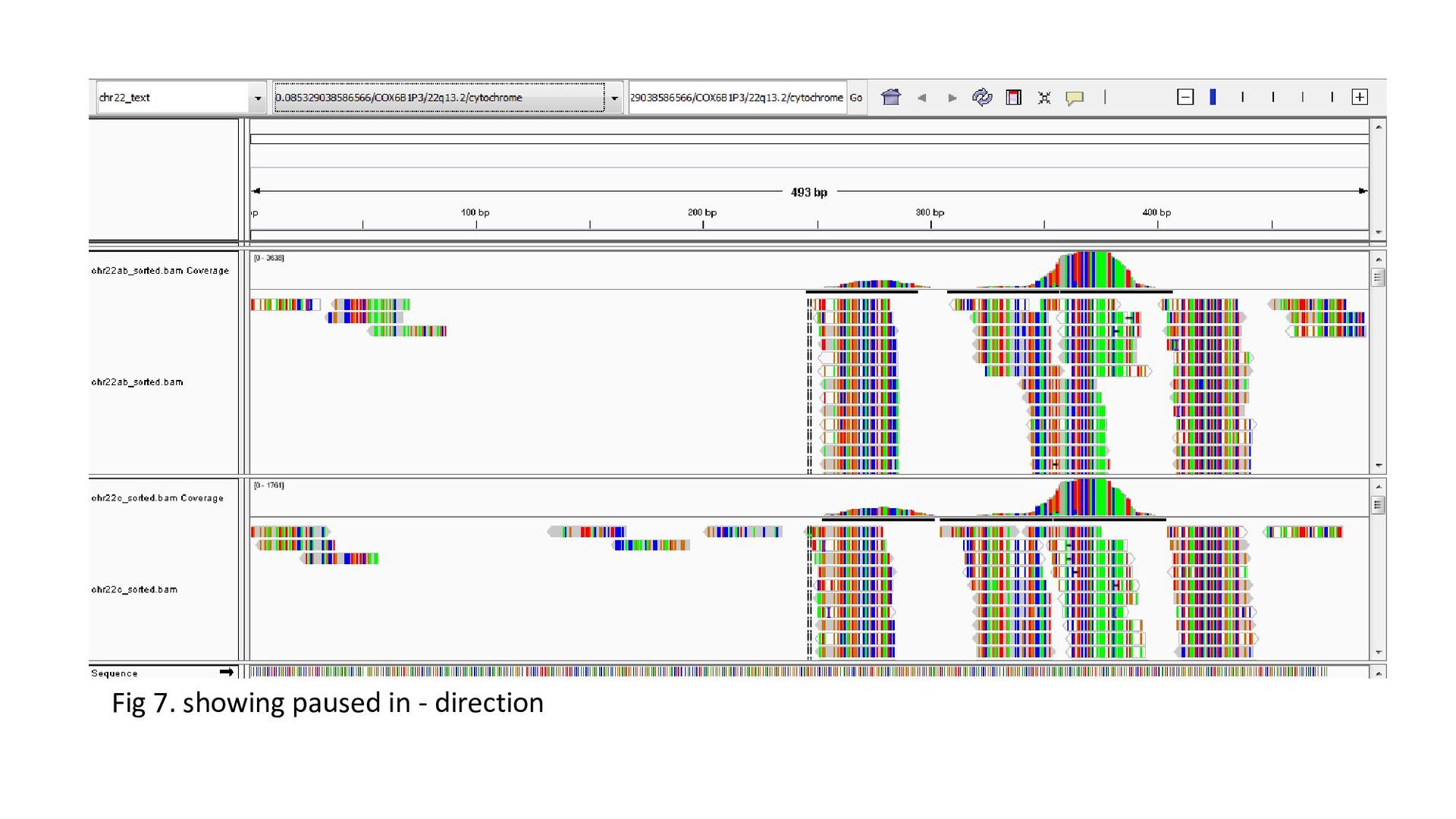
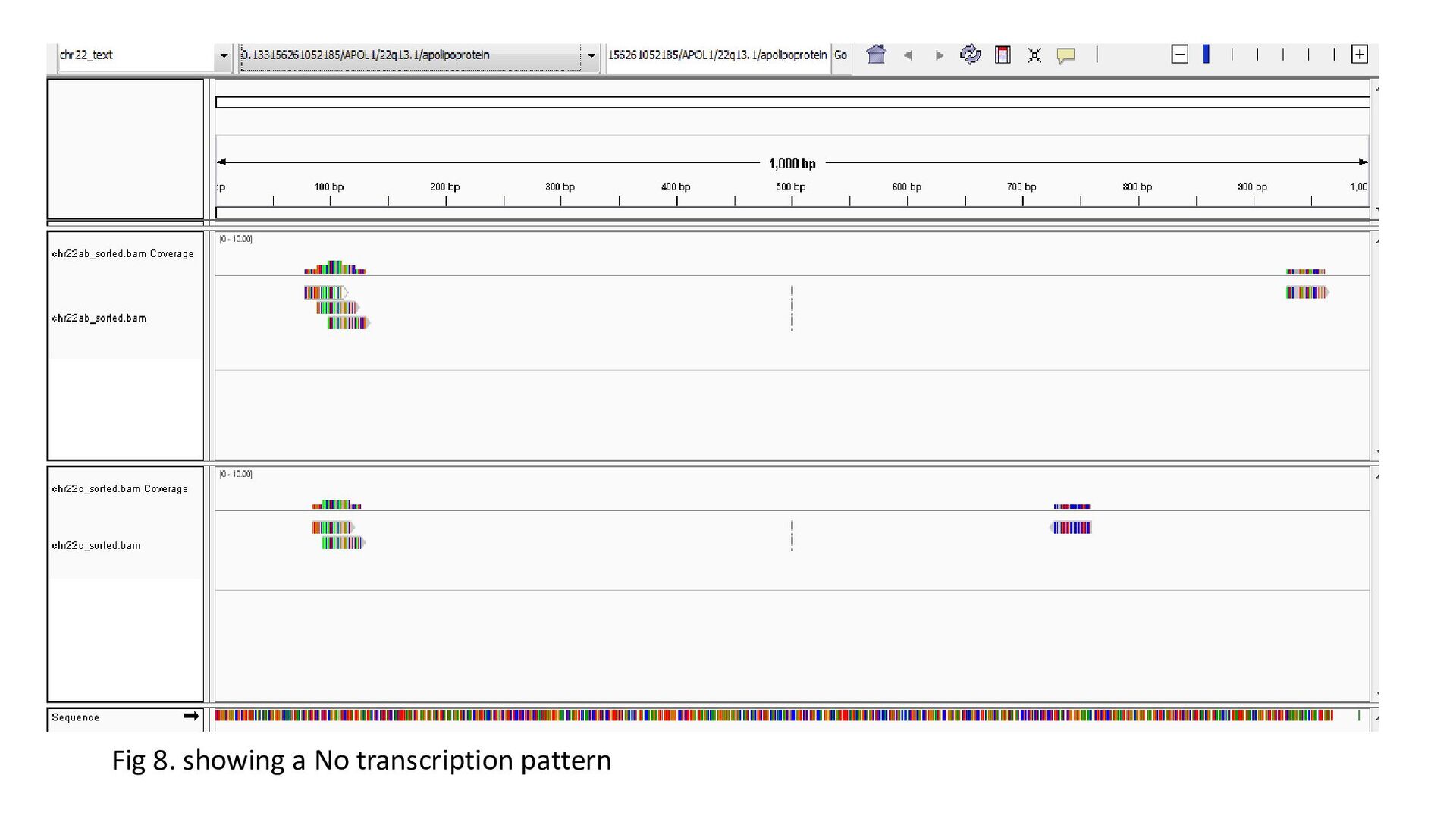
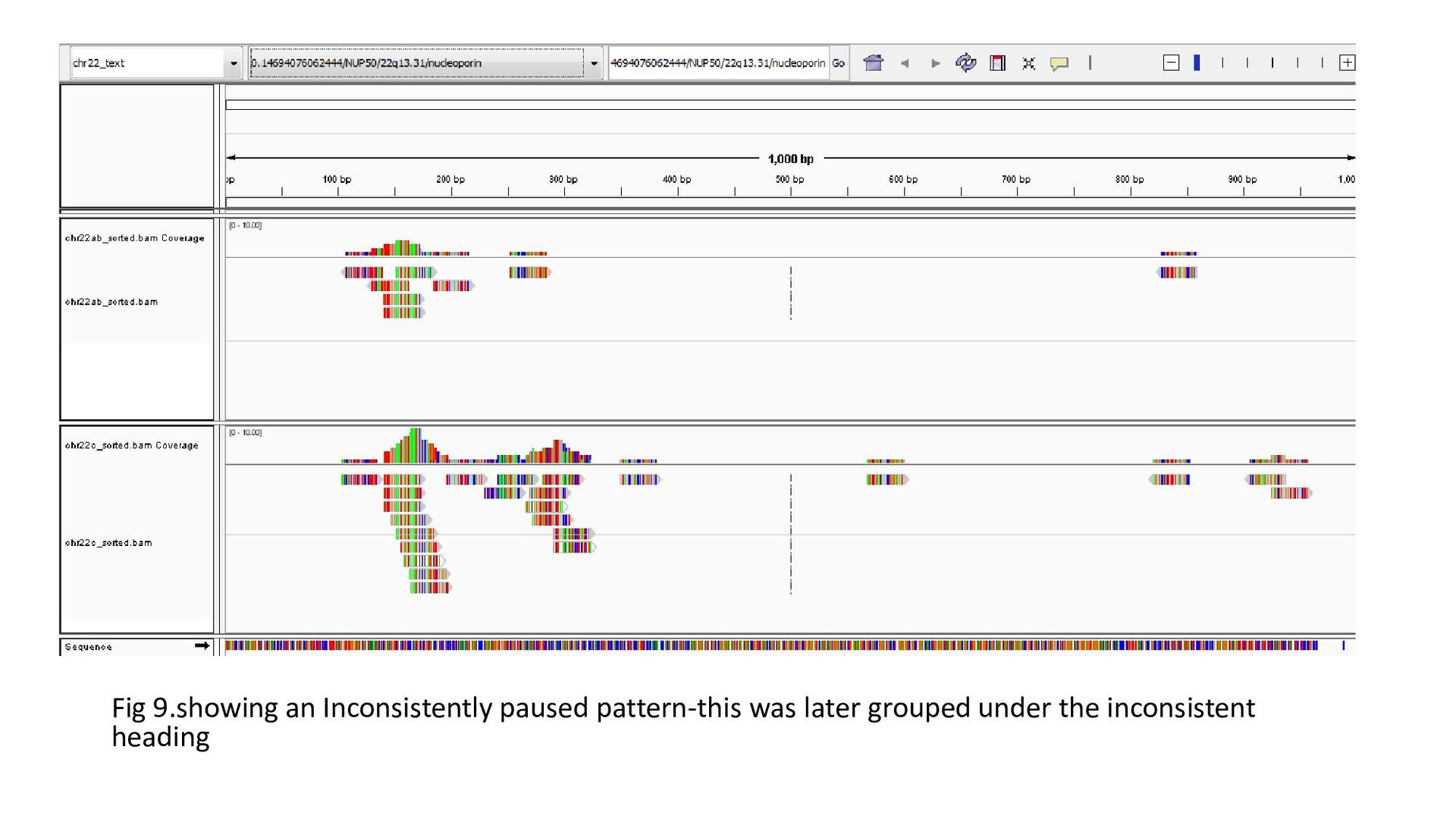
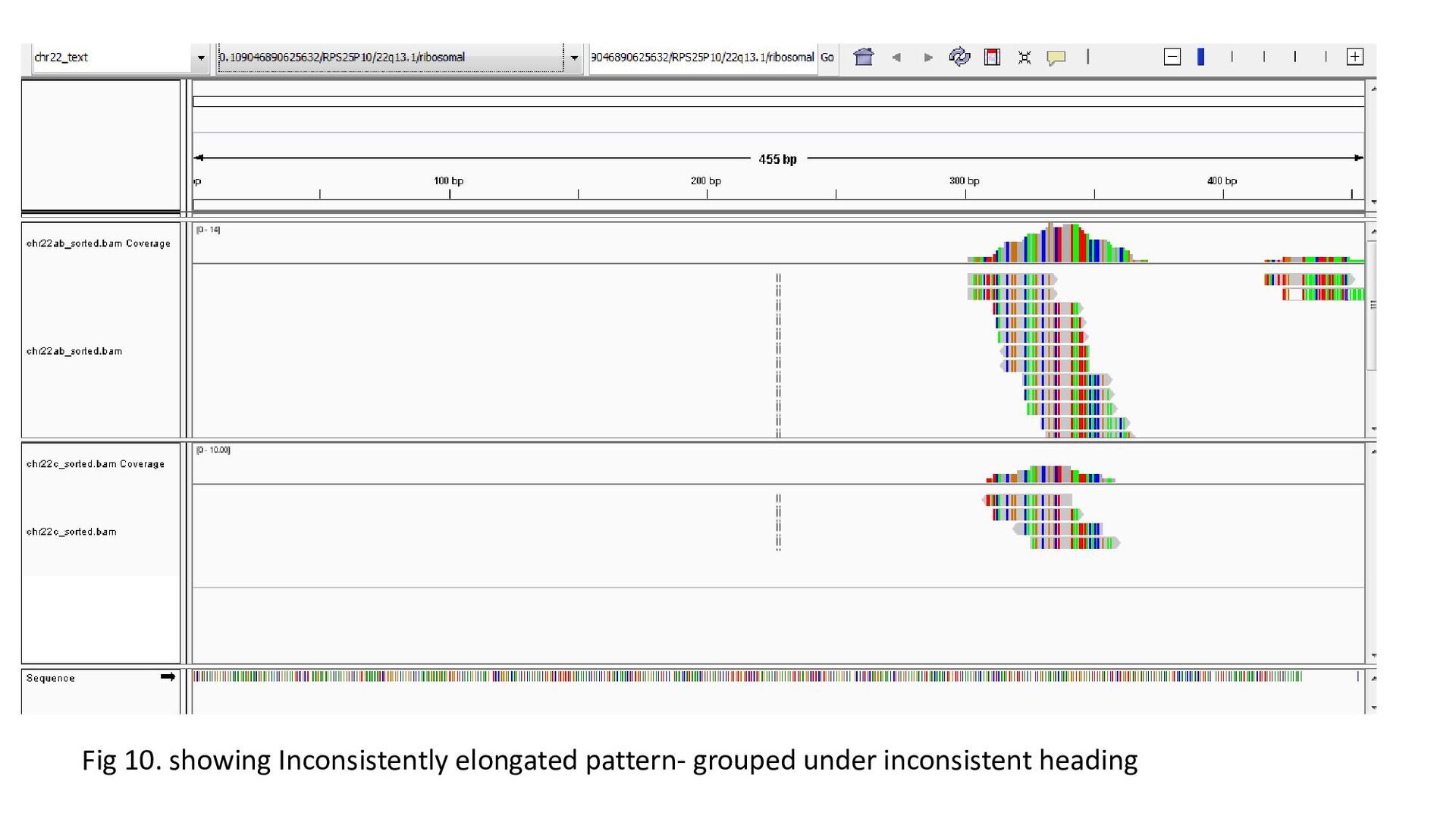
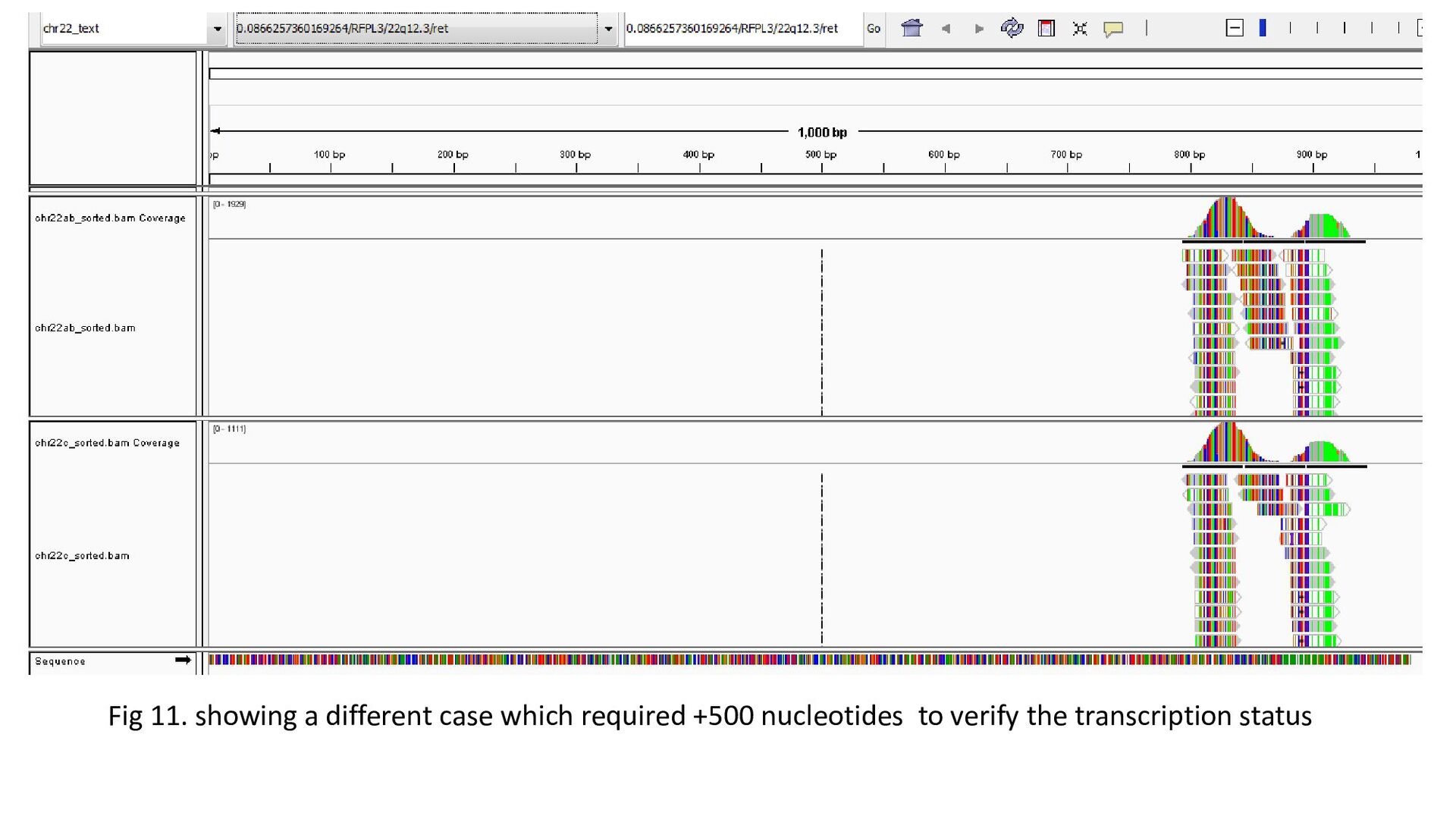
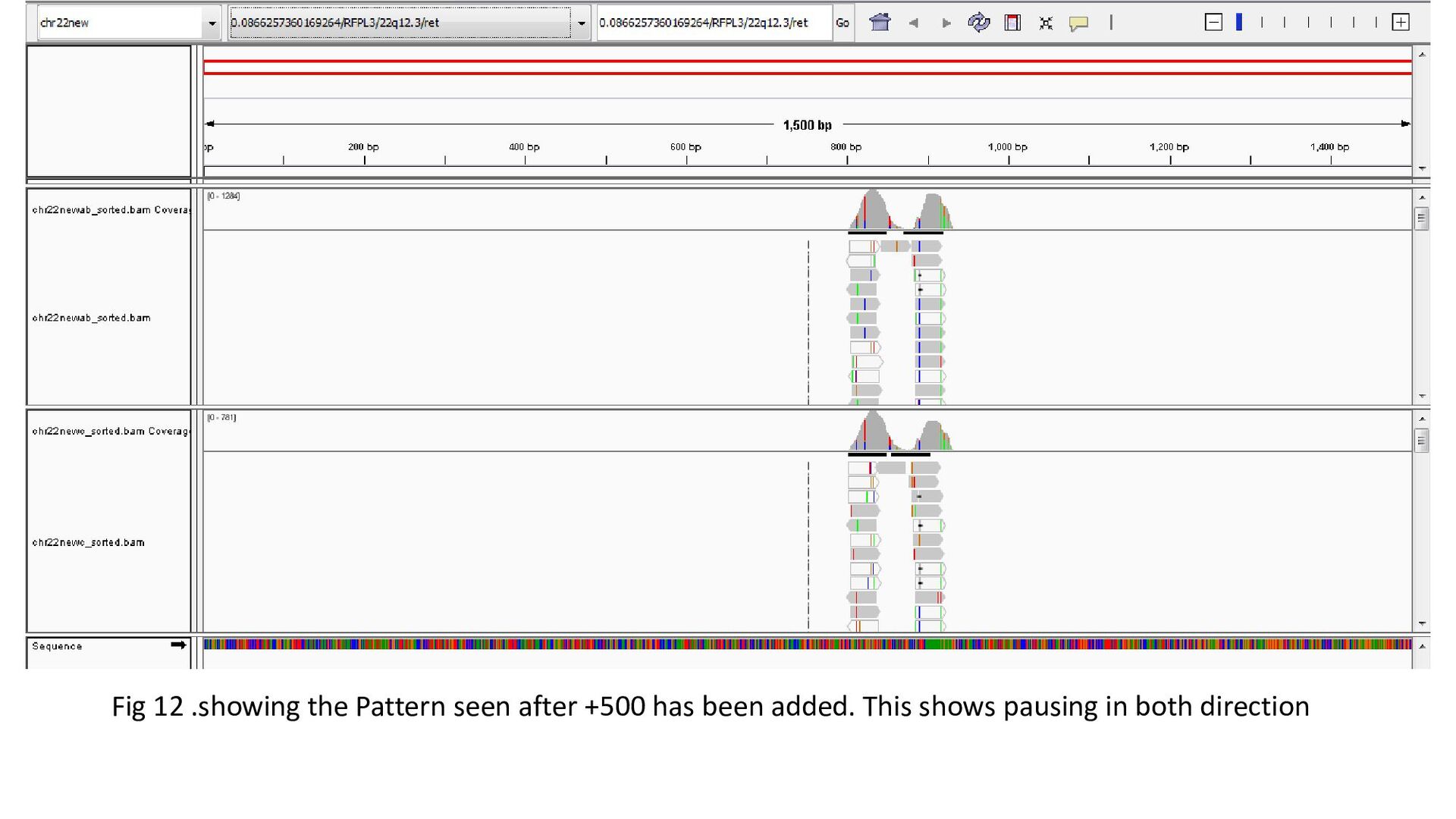
gene was characterized under 3 heads : Transcription status Bidirectional transcription Consistency • Criteria for Transcription status classifying the gene as transcriptionally elongated : no of reads is six and above in each query file, Consistent read pattern in both query files Classifying the gene as transcriptionally paused : 5X more number of reads compared to the count downstream Classifying the gene as transcriptionally silent : less than five no. of reads in both query files.
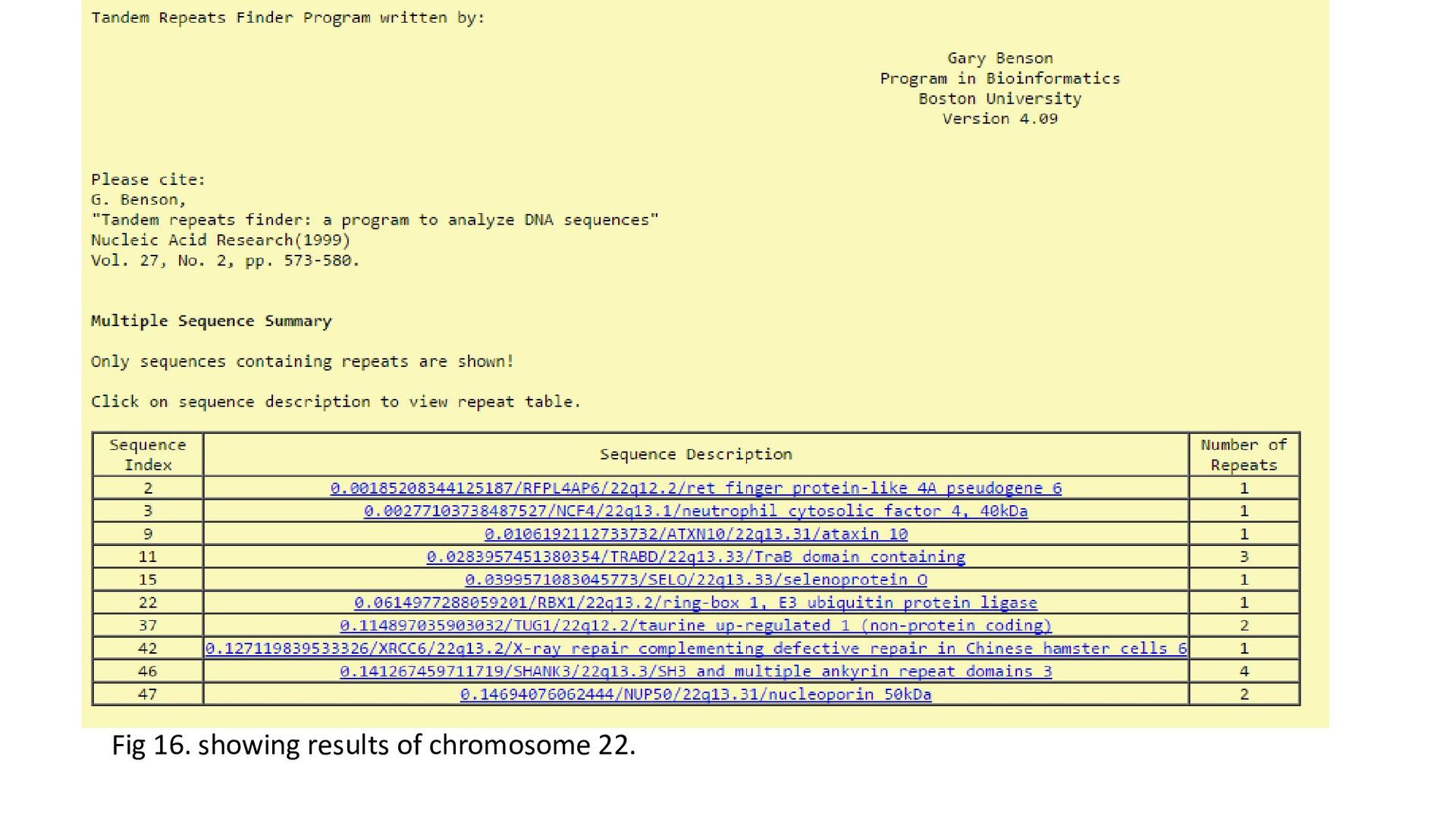
each chromosome = 50 • Inconsistent,-elongated and –paused deducted from total number to give number of genes to be analysed. • No of genes to be analysed = + elongated (+) +paused (+) elon in both dir (+) paused in both dir (+) no transcription • Number transcribed = No of genes to be analysed (-) no transcription • From the Number transcribed – number elongated and number paused was calculated • From number elongated , number elongated in + only was calculated. Likewise for paused in + • Number of TR and Rloops found .
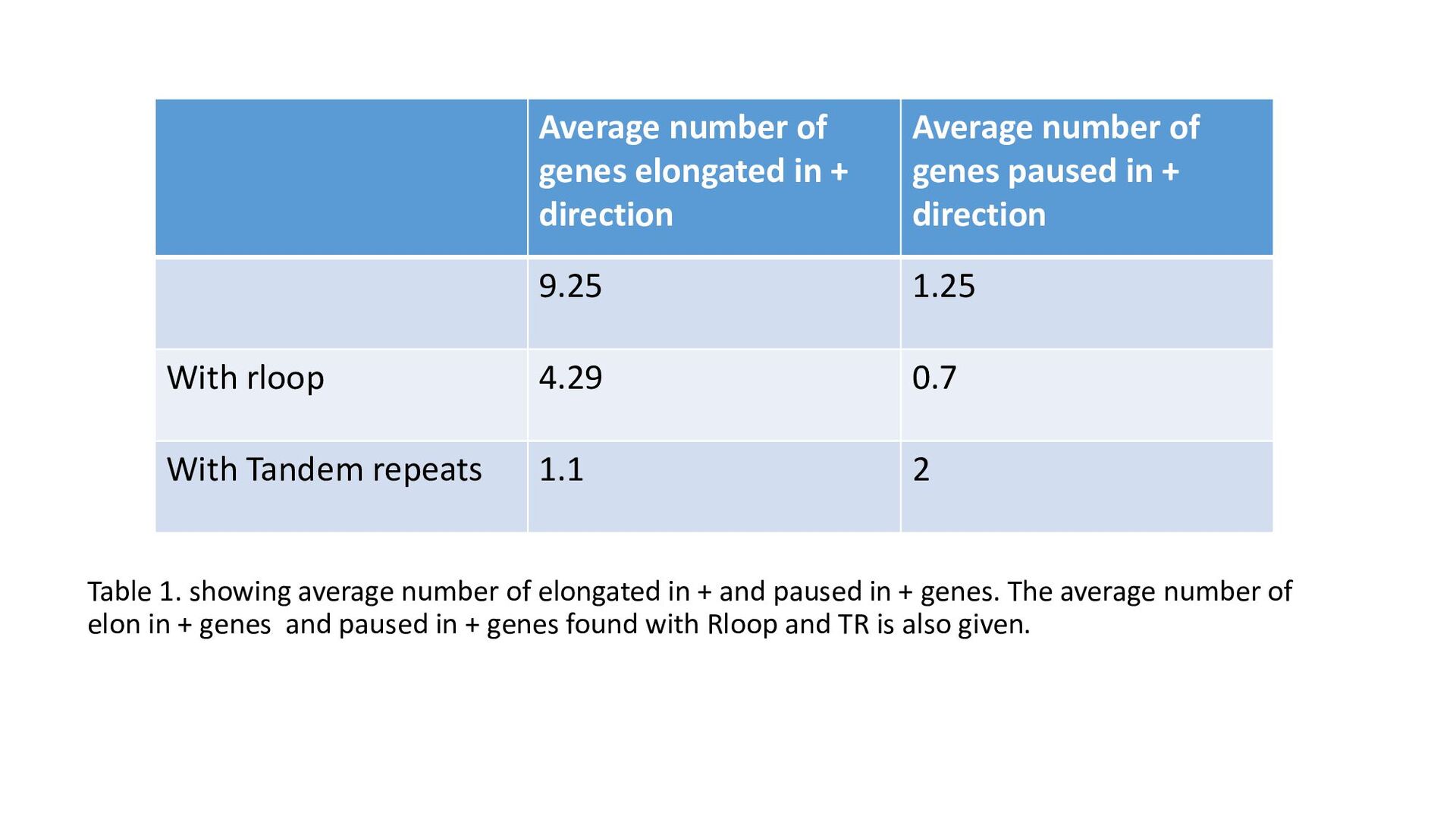
paused in + genes. The average number of elon in + genes and paused in + genes found with Rloop and TR is also given. Average number of genes elongated in + direction Average number of genes paused in + direction 9.25 1.25 With rloop 4.29 0.7 With Tandem repeats 1.1 2
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}