It happened in 1958 that John Kendrew’s group determined the three-dimensional structure of myoglobin at a resolution of 6 Å. This first view of a protein fold was a breakthrough at that time. Now, more than half a century later, both experimental and computational techniques have substantially improved as well as our understanding of how proteins and ligands interact. Yet, there are many unanswered questions to be addressed and patterns to be uncovered.
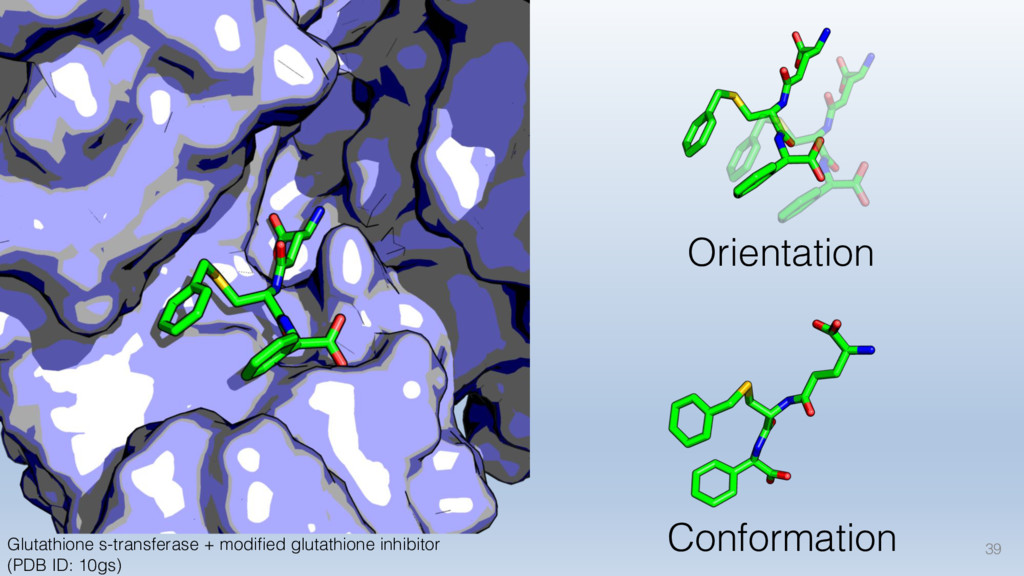
For instance, we know that precise molecular recognition is necessary for healthy, biological processes. However, the vast -- almost infinite -- number of combinations and varieties of interfacial shapes of chemical group patterns make this problem especially challenging.
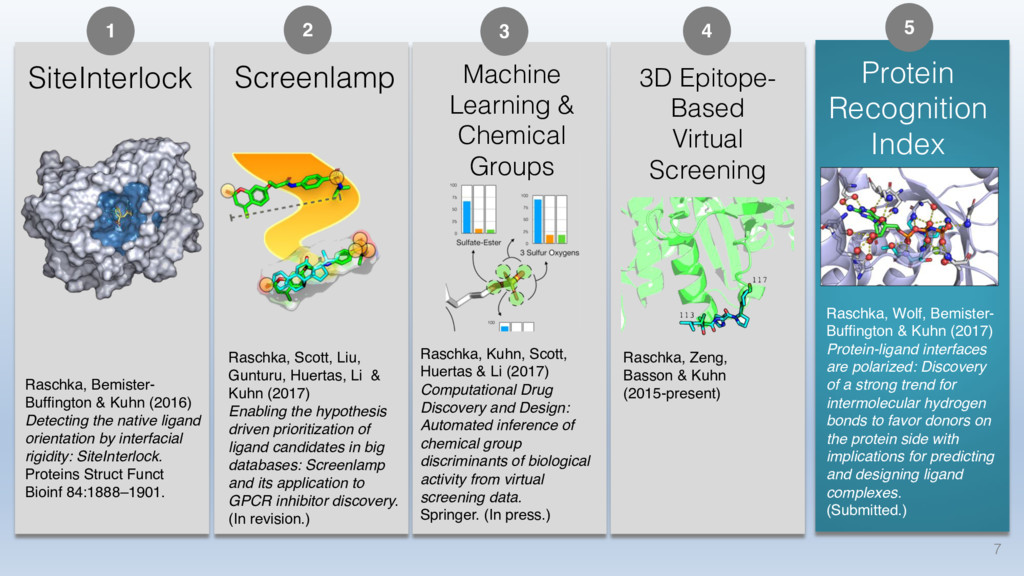
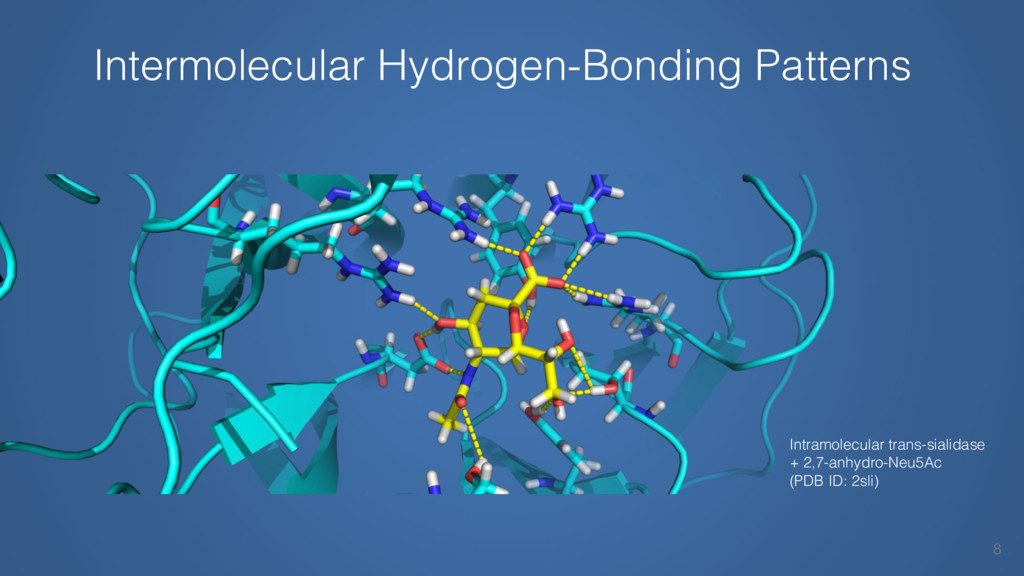
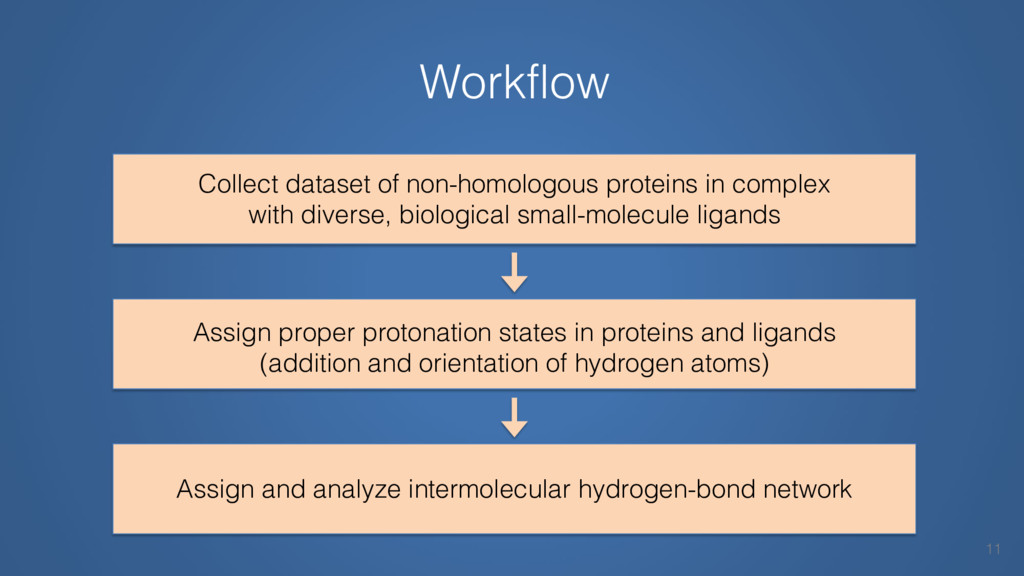
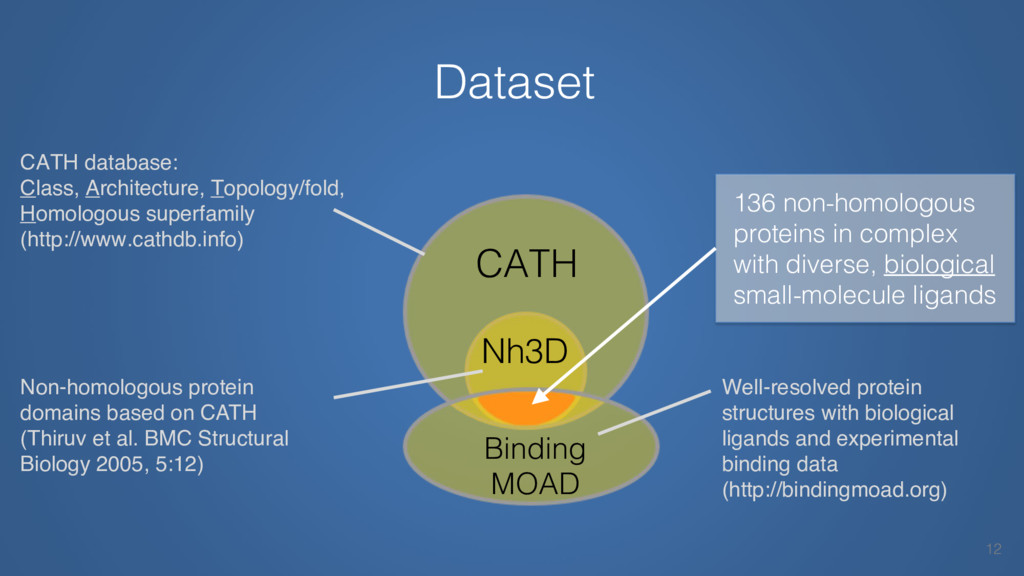
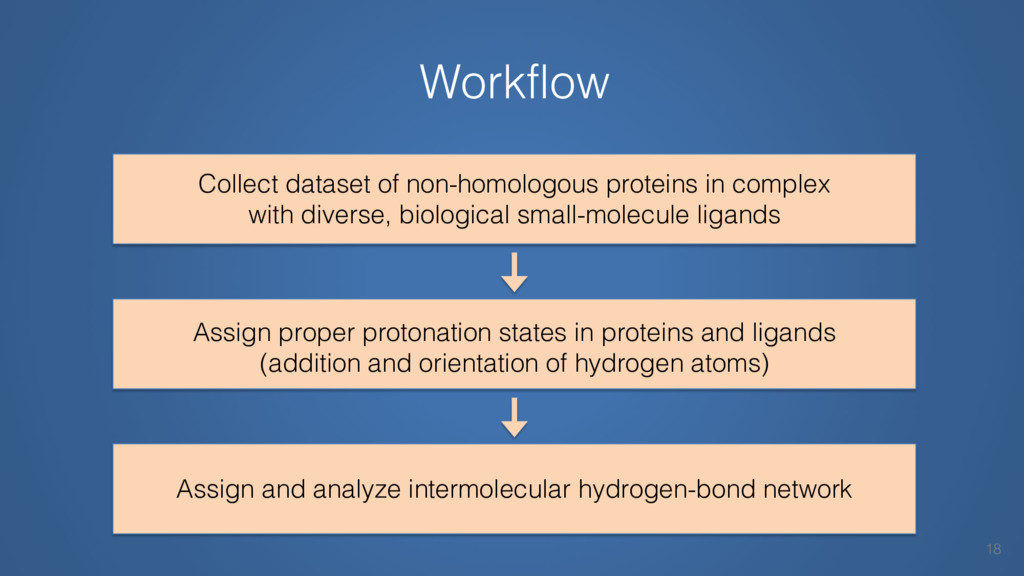
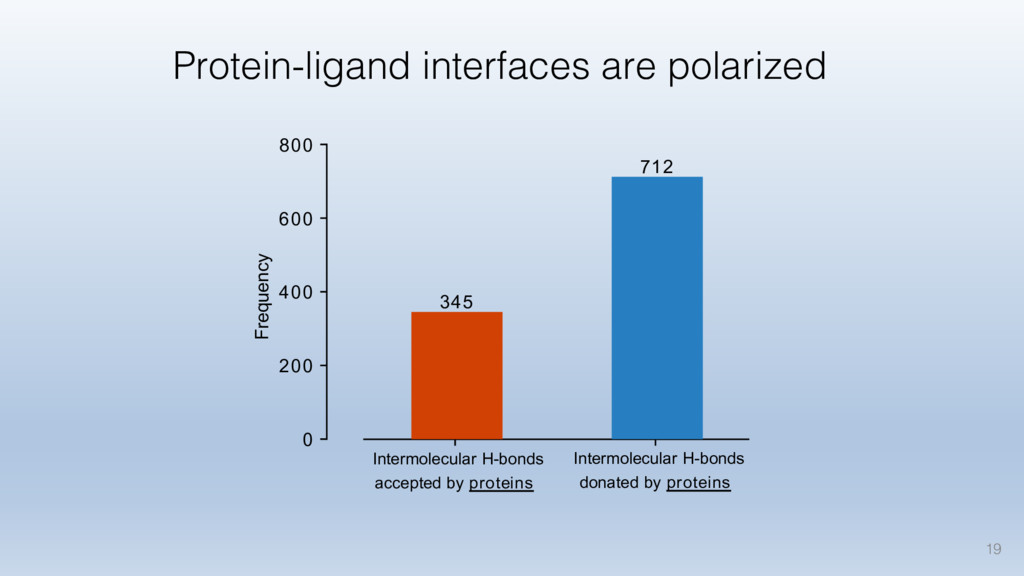
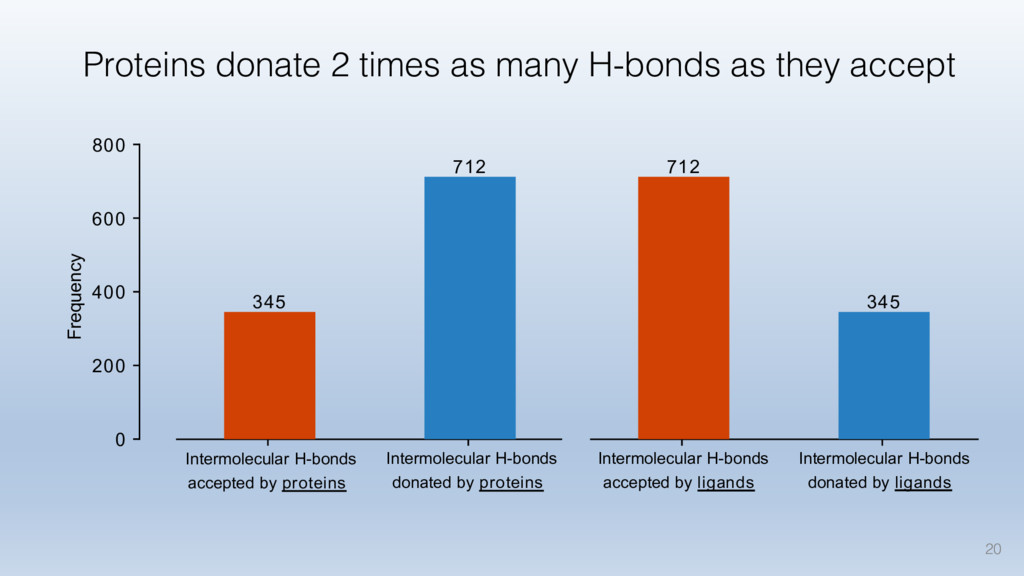
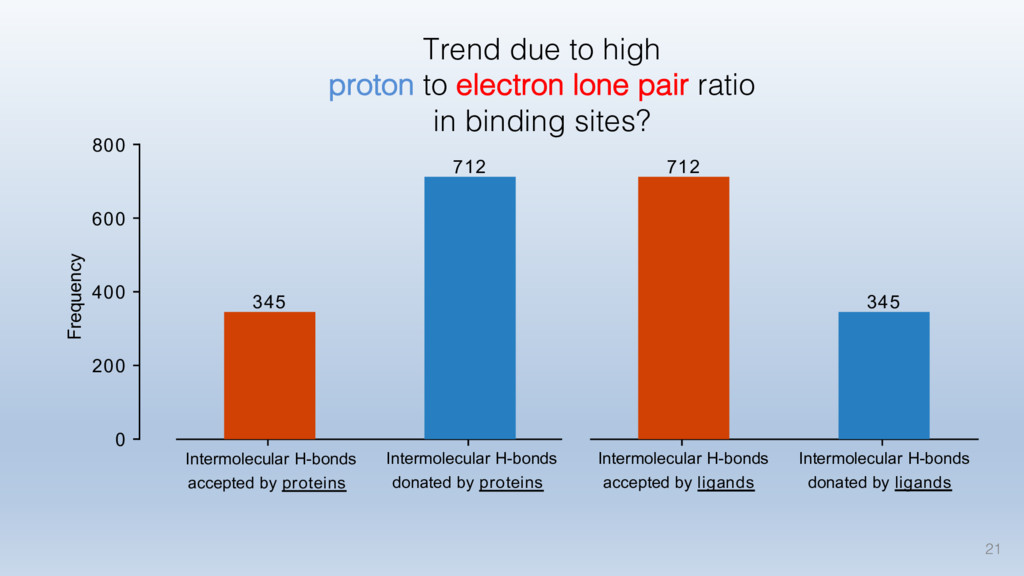
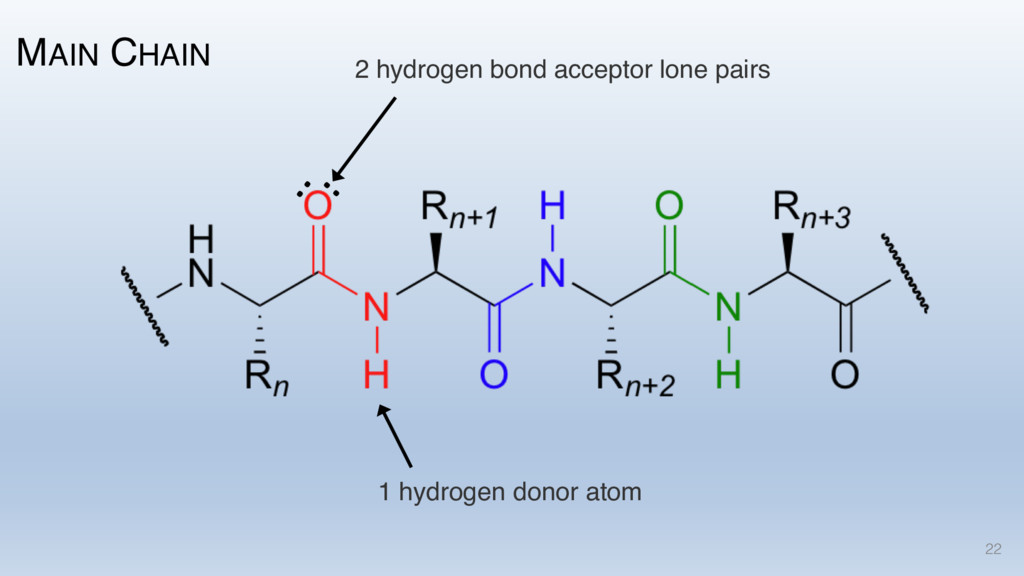
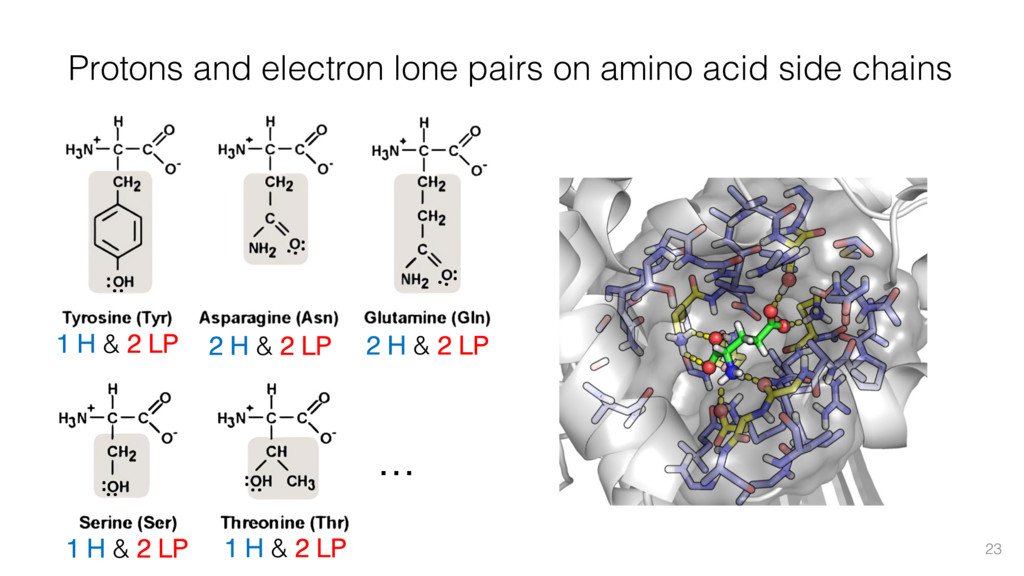
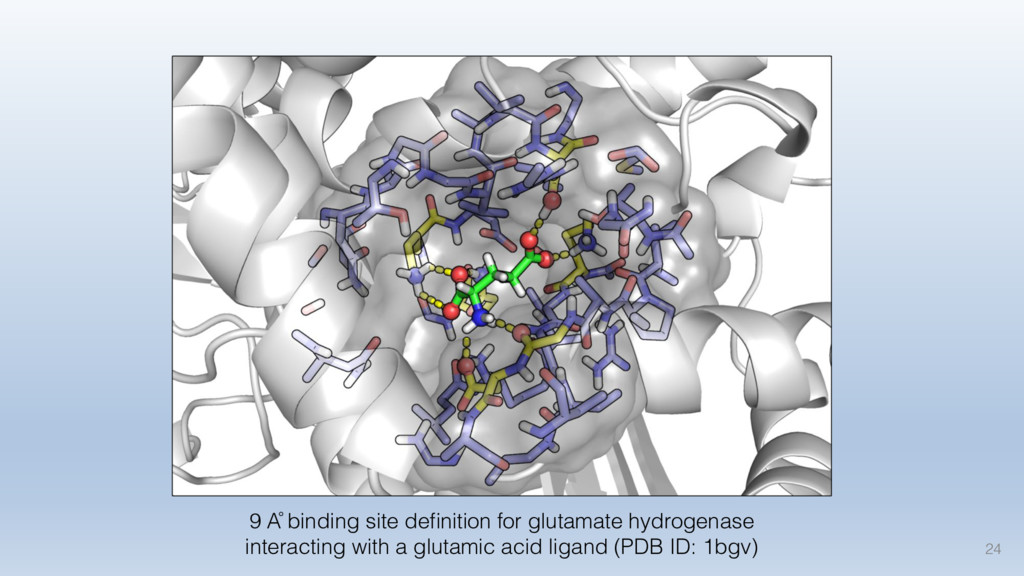
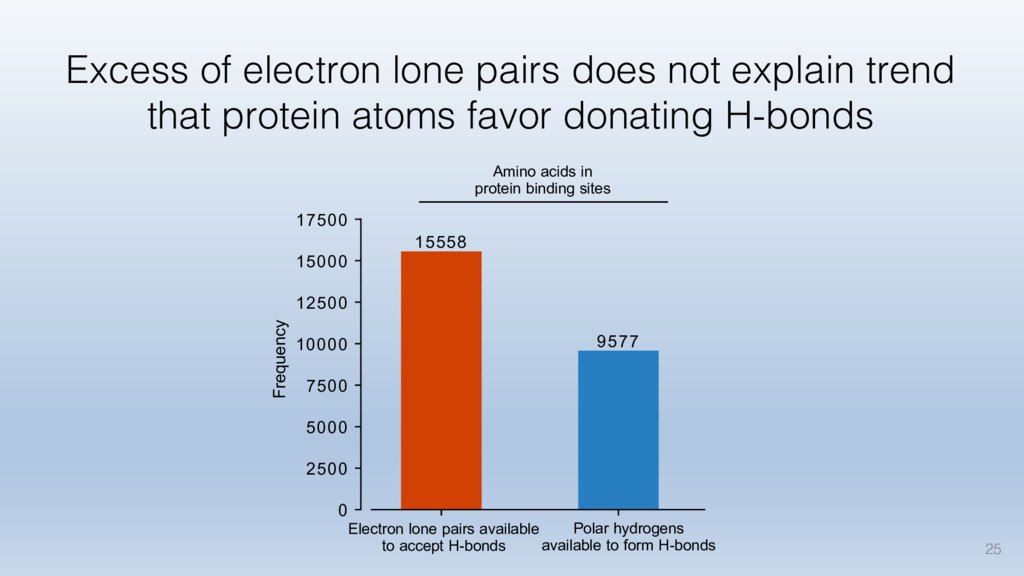
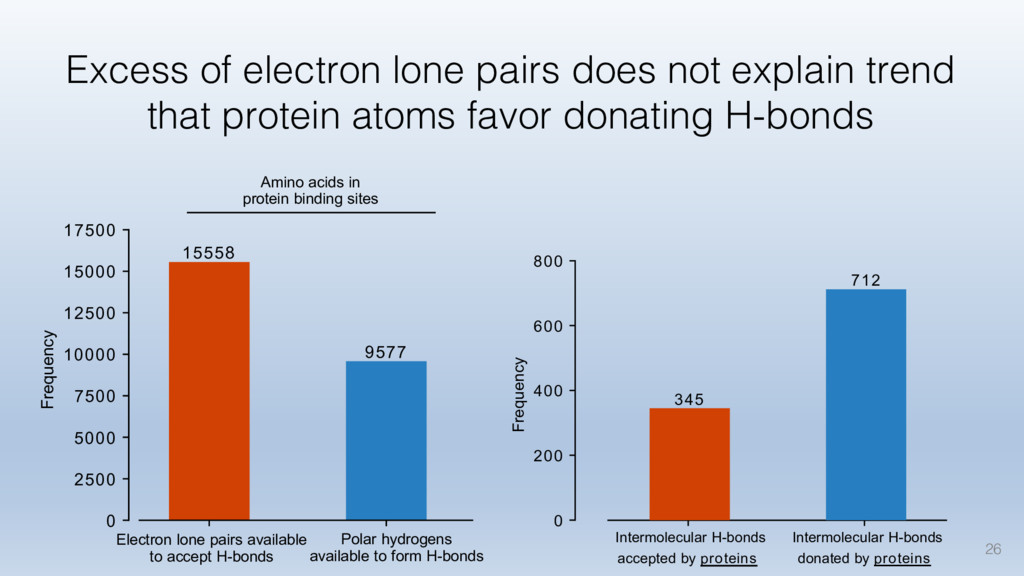
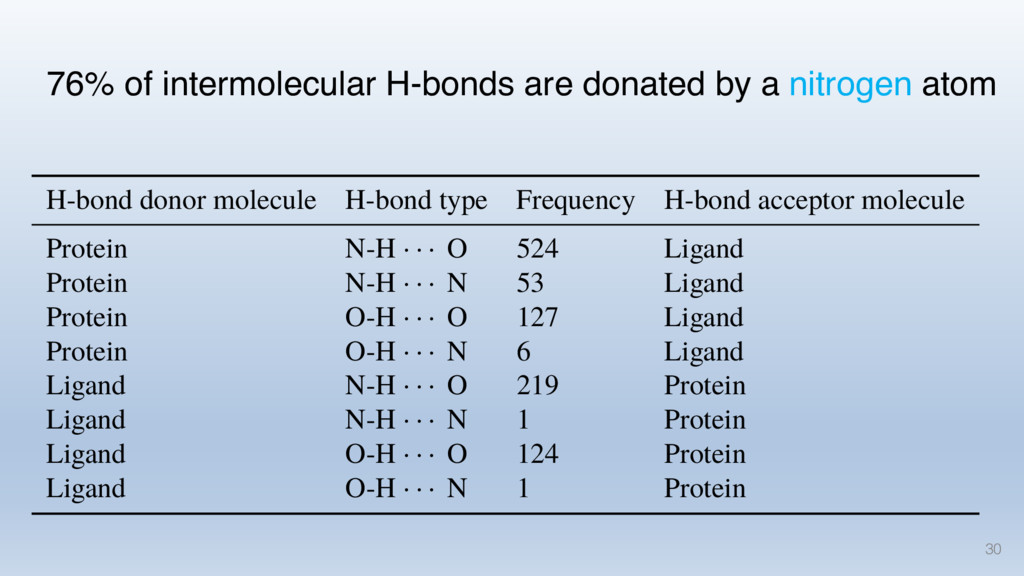
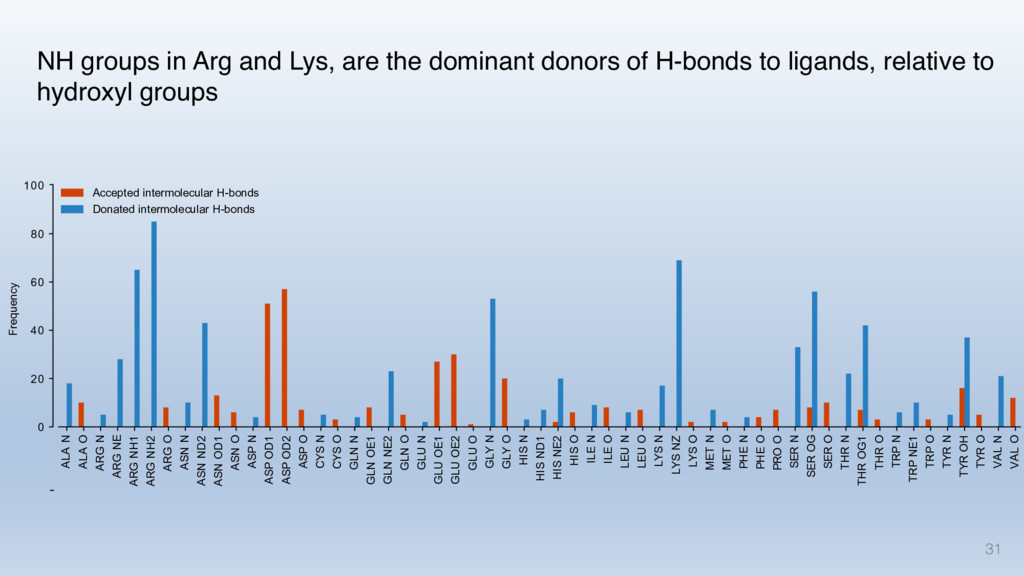
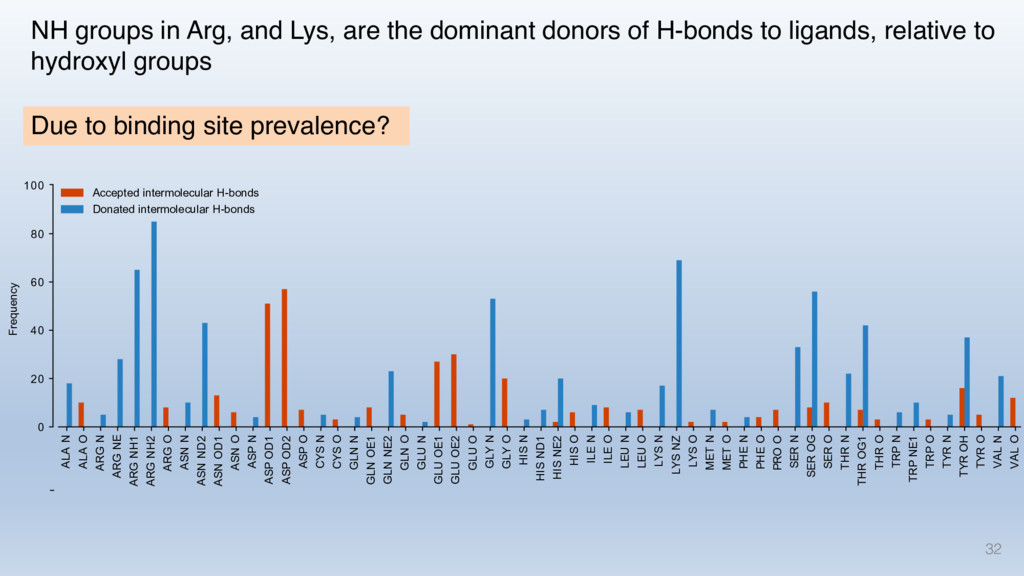
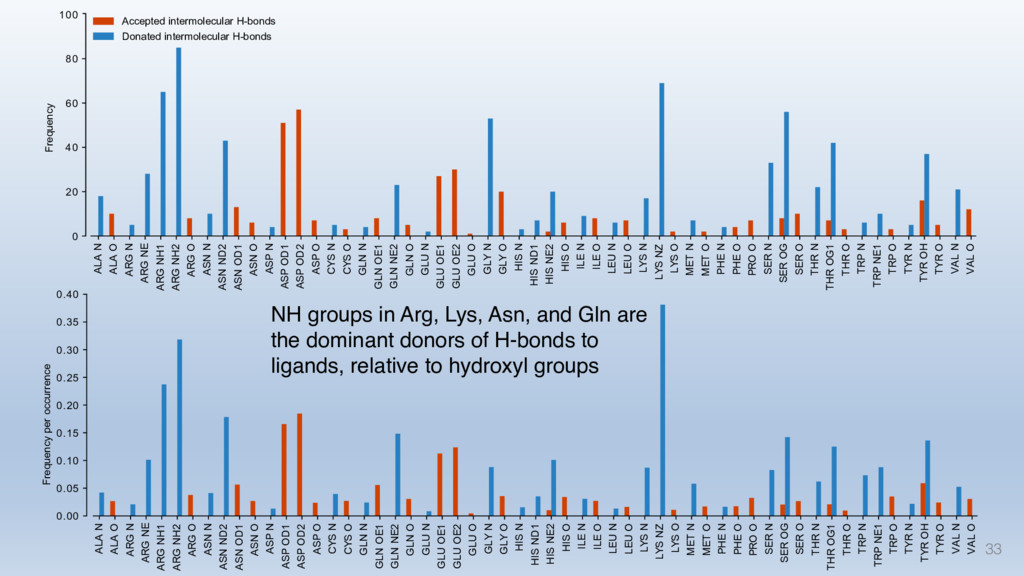
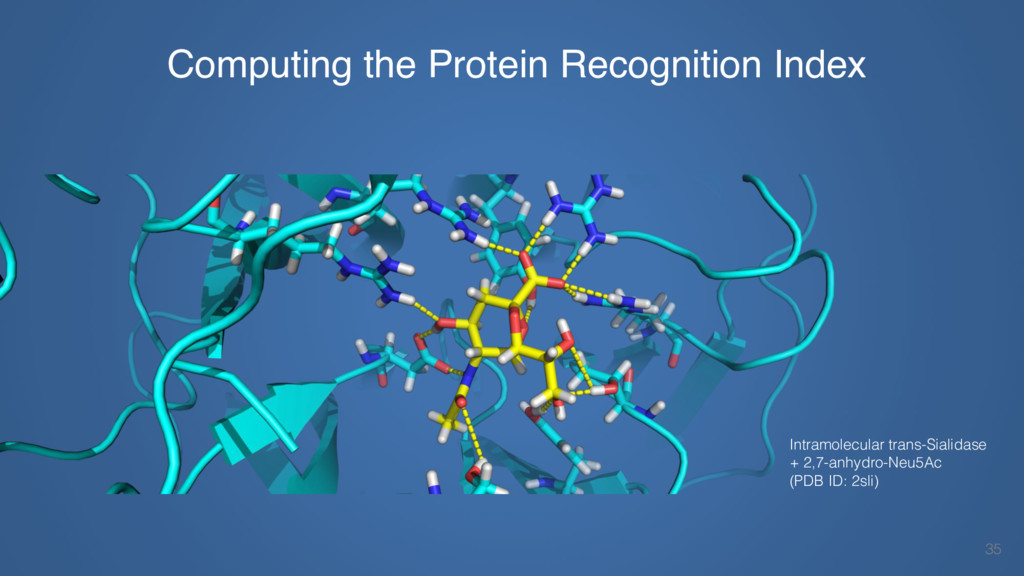
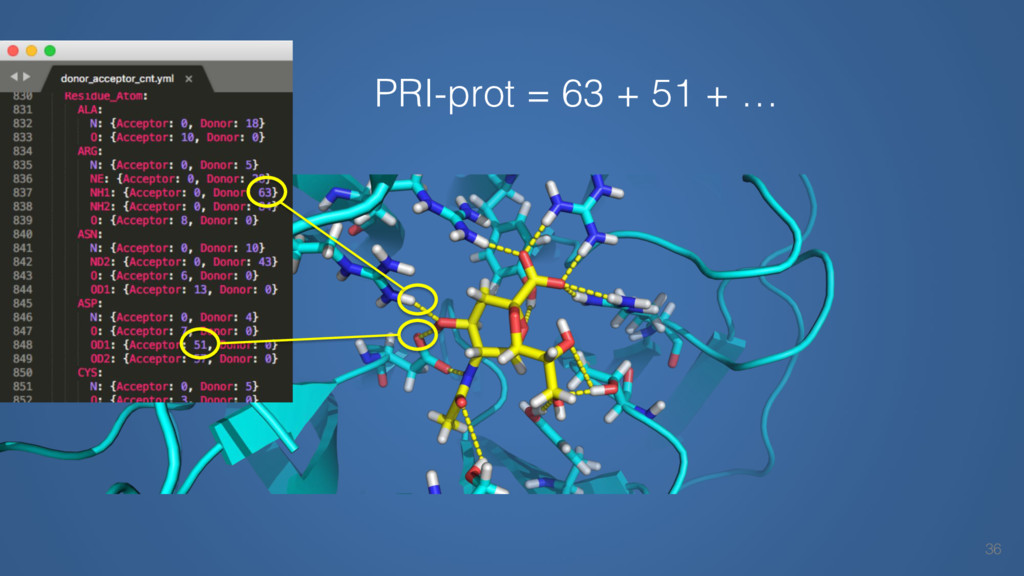
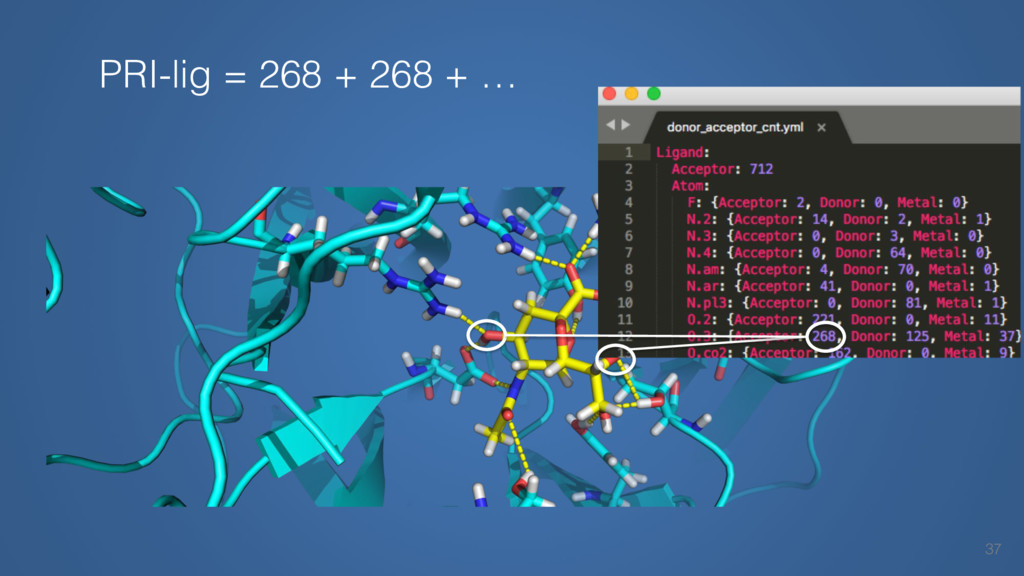
This talk will cover the analysis of a large dataset of non-homologous proteins bound to their biological ligands, to test a hypothesis that arose from observations made throughout different inhibitor discovery projects: "proteins favor donating H-bonds to ligands and avoid using groups with both H-bond donor and acceptor capacity."
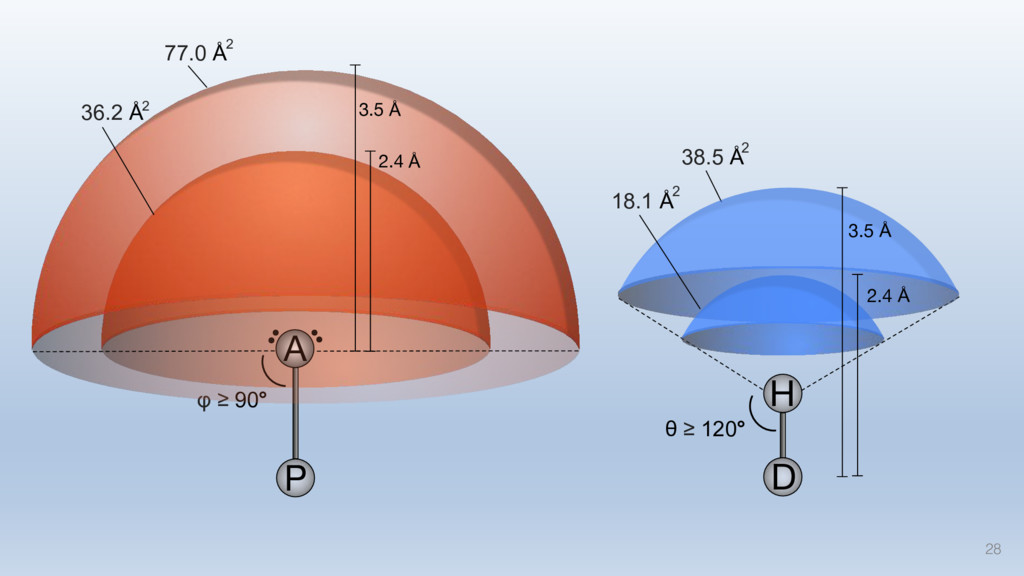
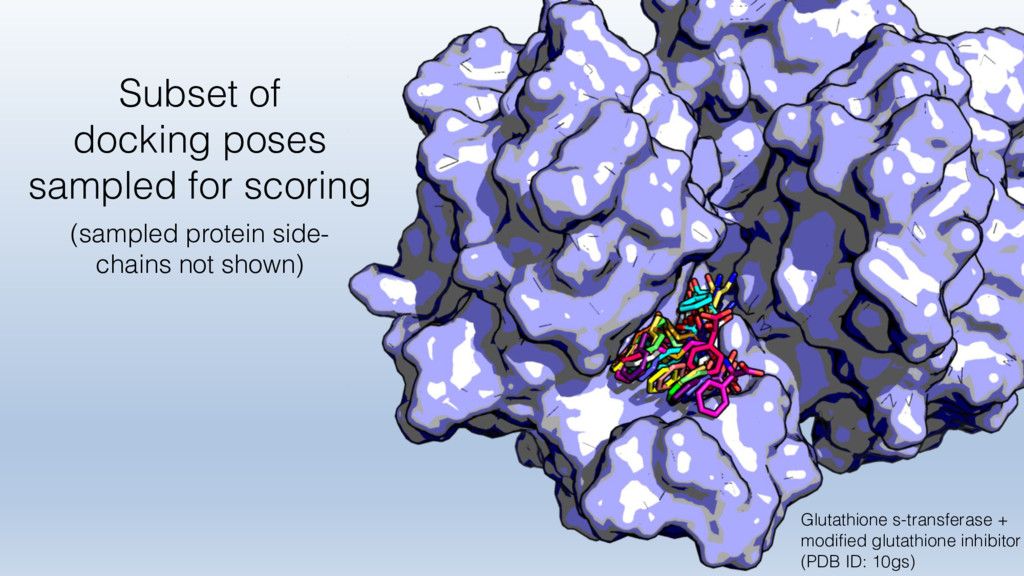
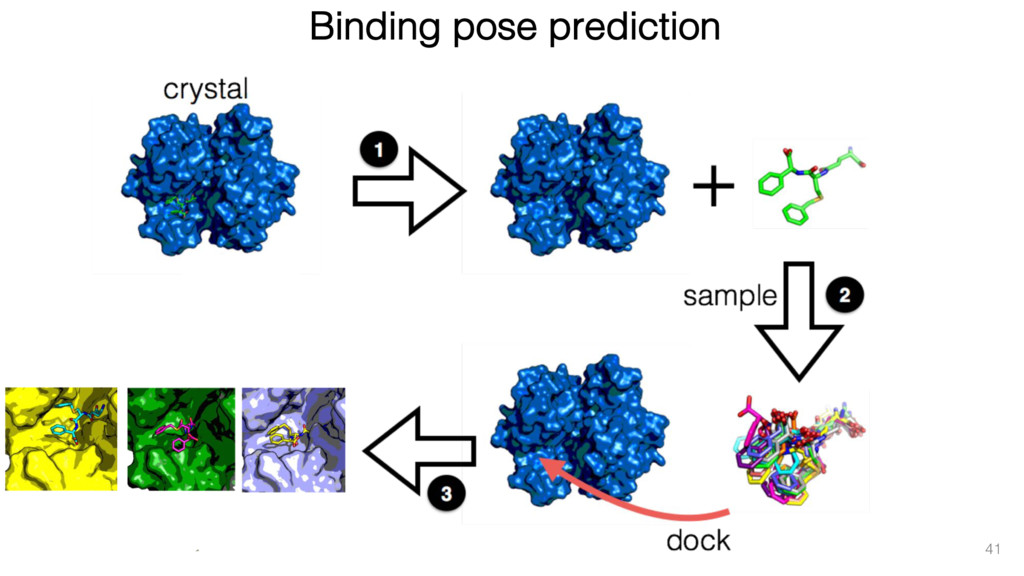
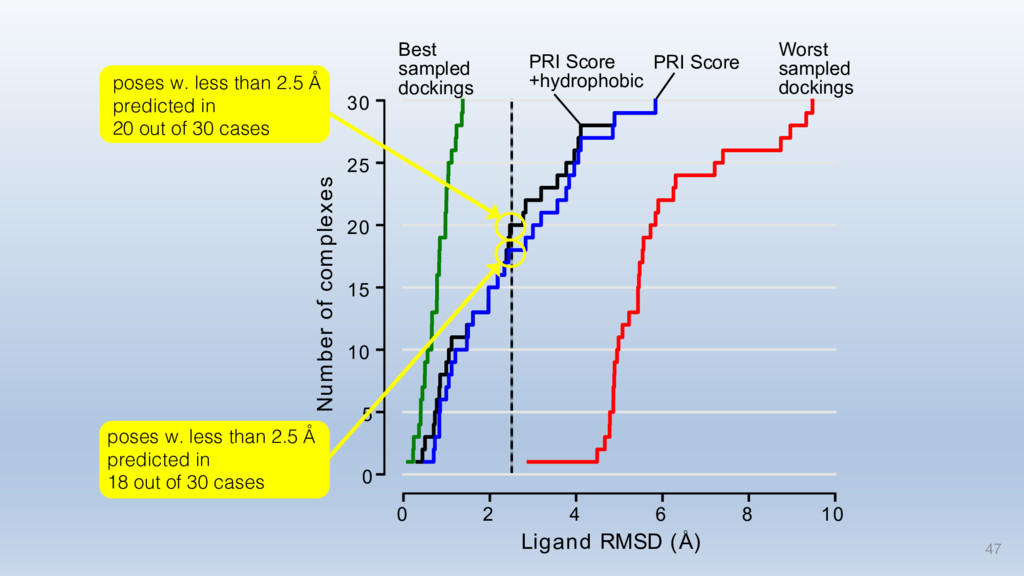
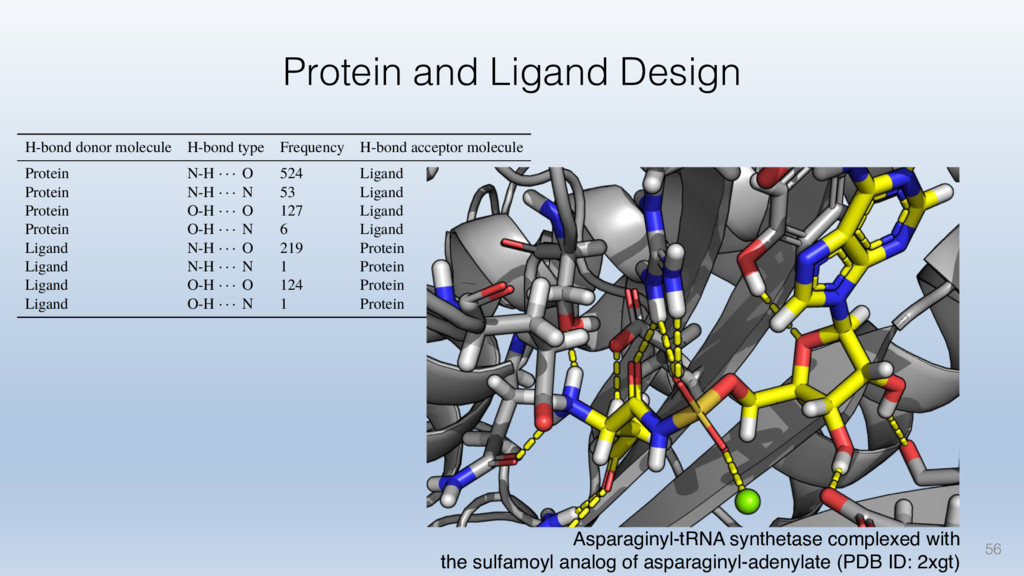
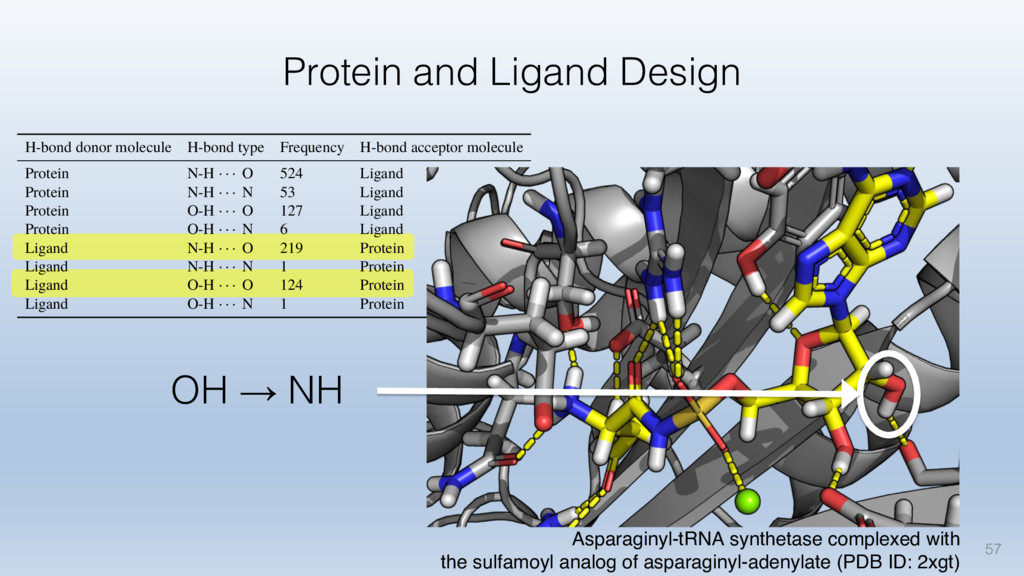
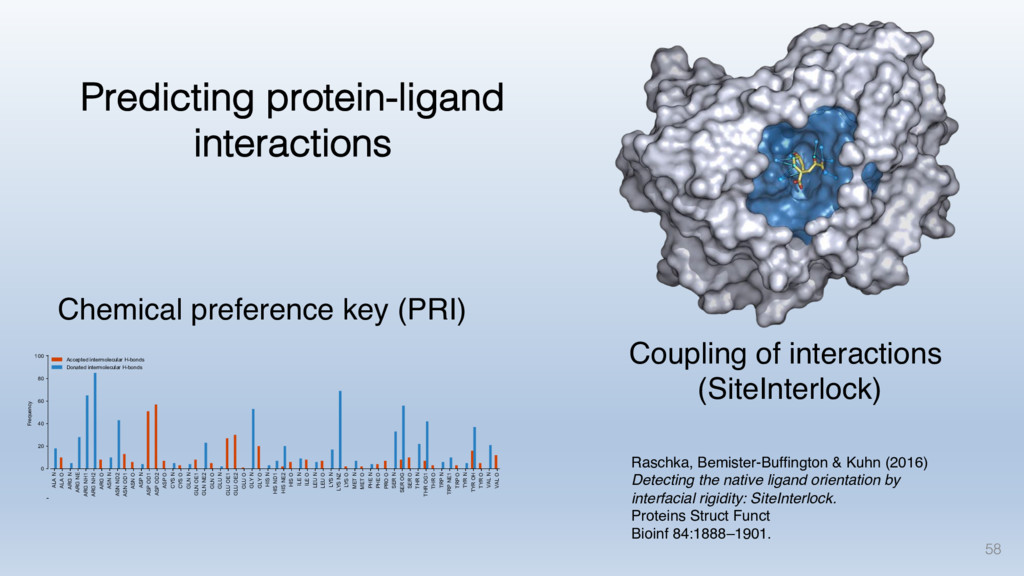
The results reveal clear and strong patterns of chemical group matching preferences for intermolecular hydrogen bonding across a database of non-homologous protein-ligand complexes. It appears that the specificity of ligands is owed to a narrow geometry when forming cognate, intermolecular H-bonds. Hence, both the chemical and geometric constraints are defining a specific hydrogen bonding pattern that is only matched by ligands with the right acceptor-rich key. Further, the results show that a preference key computed based on the patterns of chemical groups participating in H-bonding is sufficient to predict protein-ligand complexes. Finally, the trends observed in this study can guide ligand design and protein mutagenesis in studies of protein-ligand interactions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}