genomic data analysis and collaboration. Nat Rev Genet. 2018 May;19(5):325. https://doi.org/10.1038/nrg.2017.113 • Wilks C, Gaddipati P, Nellore A, Langmead B. Snaptron: querying splicing patterns across tens of thousands of RNA-seq samples. Bioinformatics. 2018 Jan 1;34(1):114-116. https:// doi.org/10.1093/bioinformatics/btx547 • Collado-Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD, Jaffe AE, Langmead B, Leek JT. Reproducible RNA-seq analysis using recount2. Nat Biotechnol. 2017 Apr 11;35(4): 319-321. https://doi.org/10.1038/nbt.3838 • Nellore A, Jaffe AE, Fortin JP, Alquicira-Hernández J, Collado-Torres L, Wang S, Phillips RA III, Karbhari N, Hansen KD, Langmead B, Leek JT. Human splicing diversity and the extent of unannotated splice junctions across human RNA-seq samples on the Sequence Read Archive. Genome Biol. 2016 Dec 30;17(1):266. https://doi.org/10.1186/s13059-016-1118-6 • Nellore A, Collado-Torres L, Jaffe AE, Alquicira-Hernández J, Wilks C, Pritt J, Morton J, Leek JT, Langmead B. Rail-RNA: scalable analysis of RNA-seq splicing and coverage. Bioinformatics. 2017 Dec 15;33(24):4033-4040. https://doi.org/10.1093/bioinformatics/btw575
![Ben Langmead Assistant Professor, JHU Computer Science [email protected], langmead-lab.org, @BenLangmead](https://files.speakerdeck.com/presentations/c196465ed03846ffb69a69dba80aa216/slide_0.jpg){kind=link}
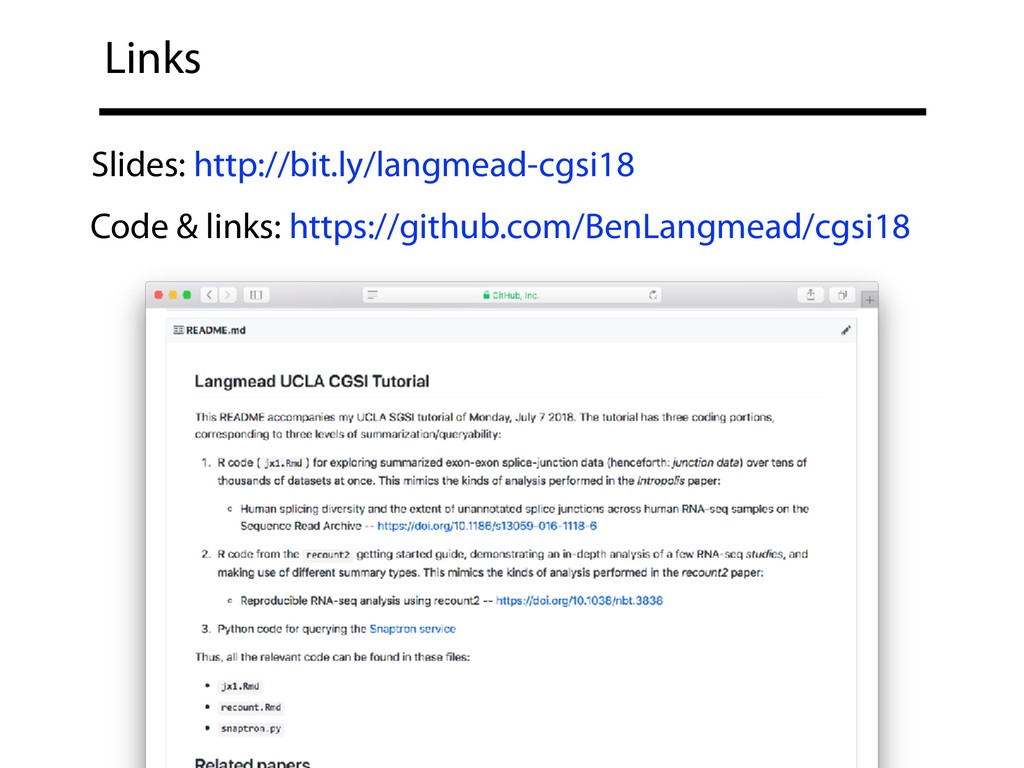{kind=link}
{kind=link}
{kind=link}
{kind=link}
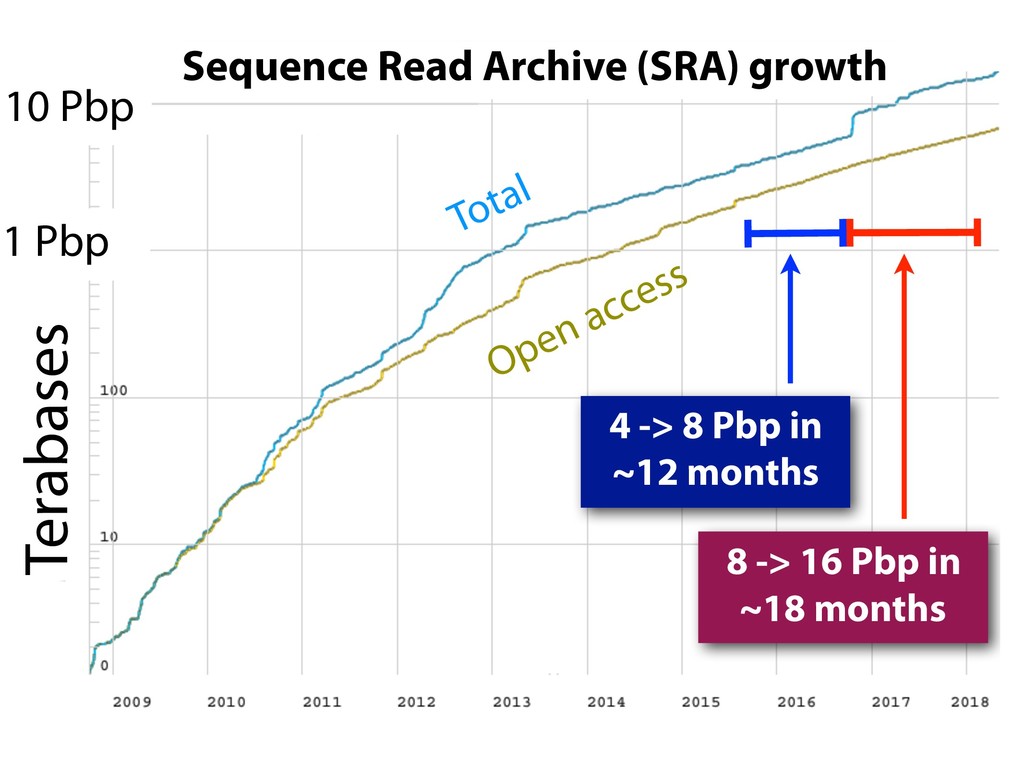{kind=link}
{kind=link}
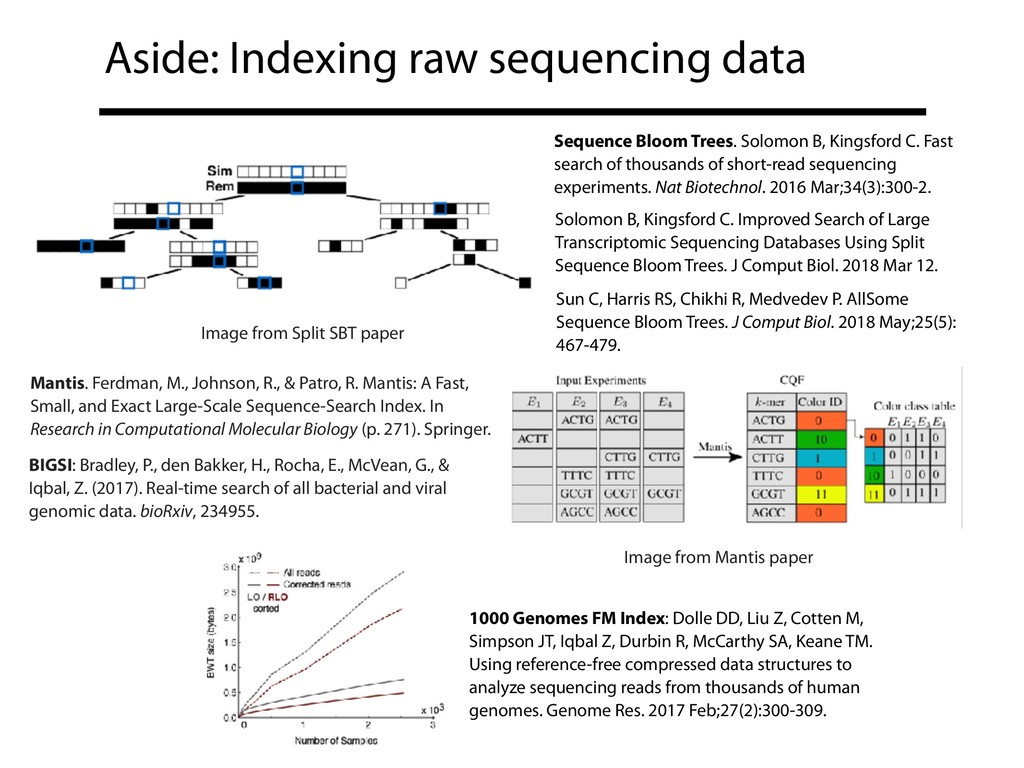{kind=link}
{kind=link}
{kind=link}
{kind=link}
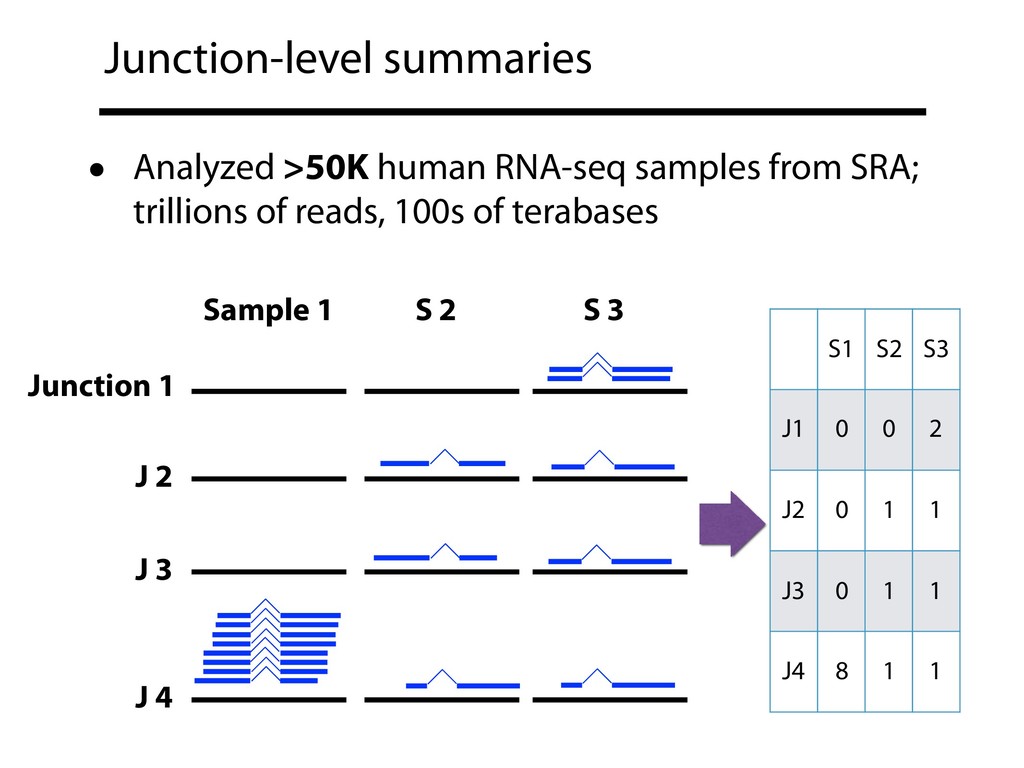{kind=link}
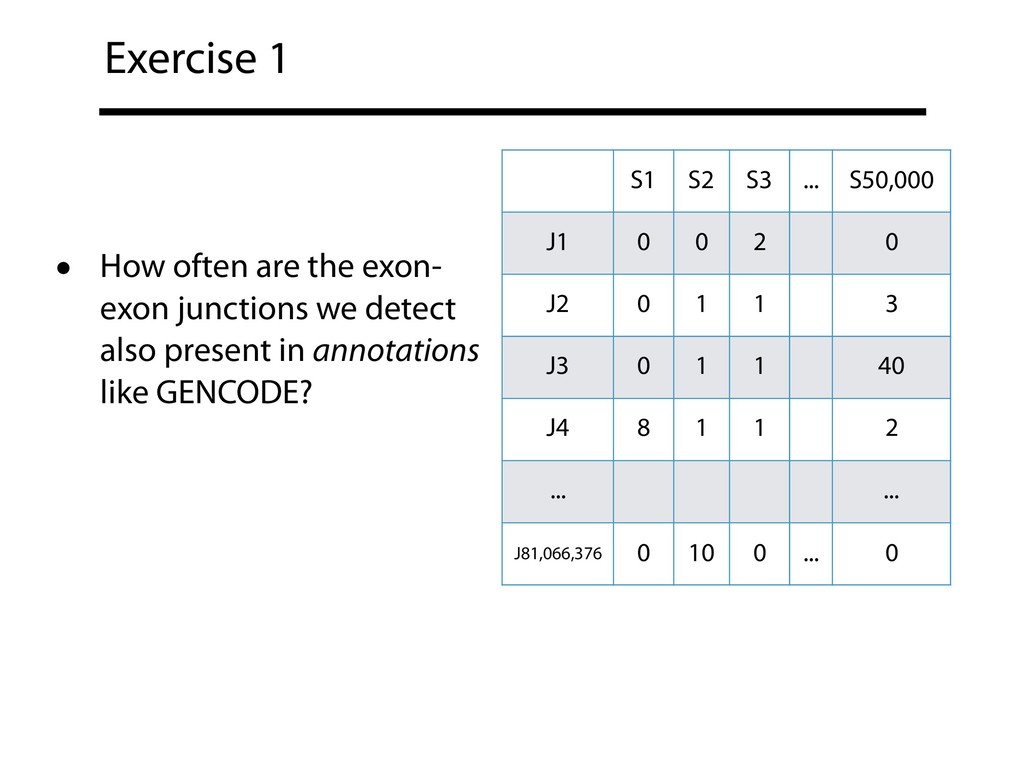{kind=link}
{kind=link}
{kind=link}
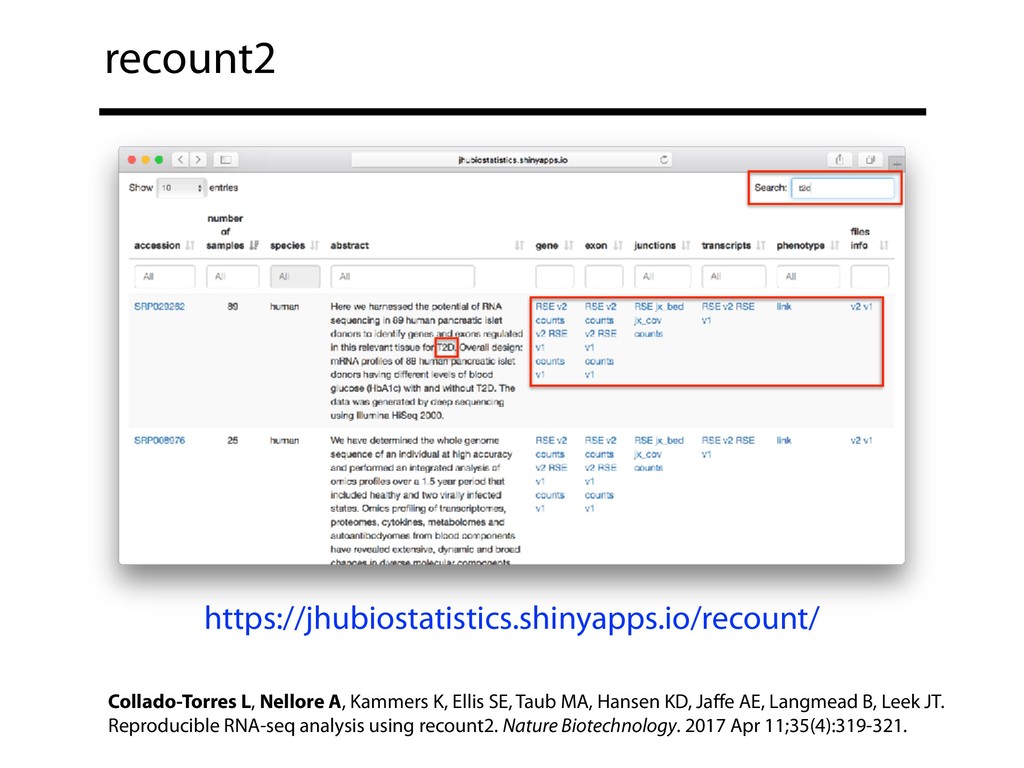{kind=link}
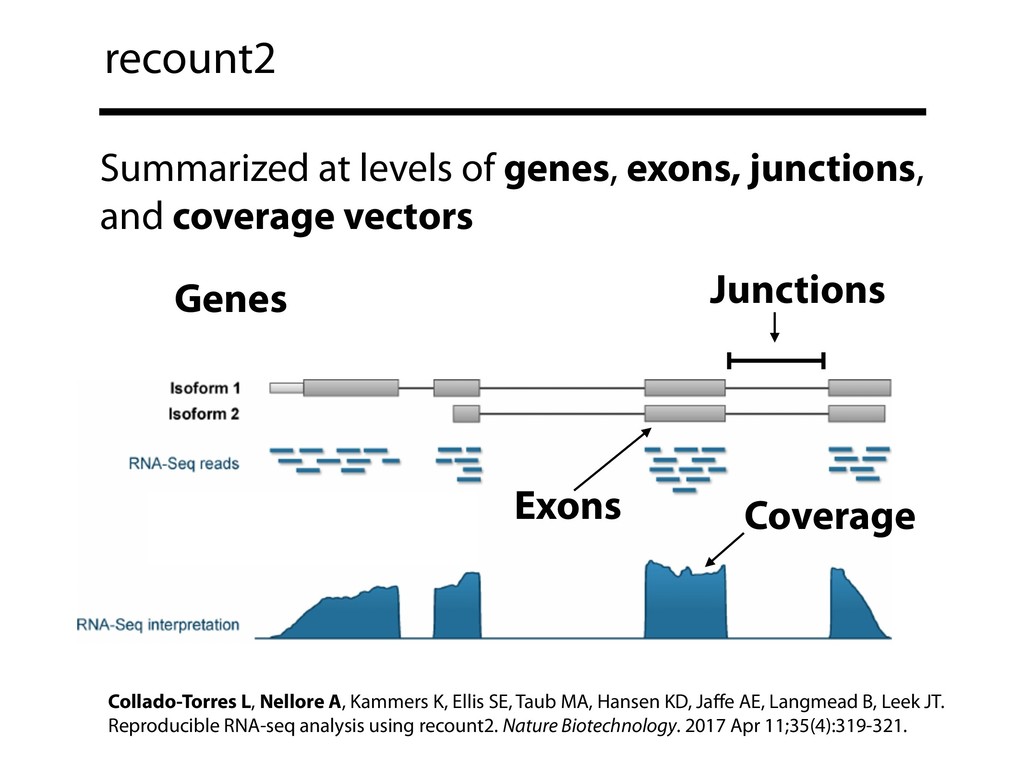{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
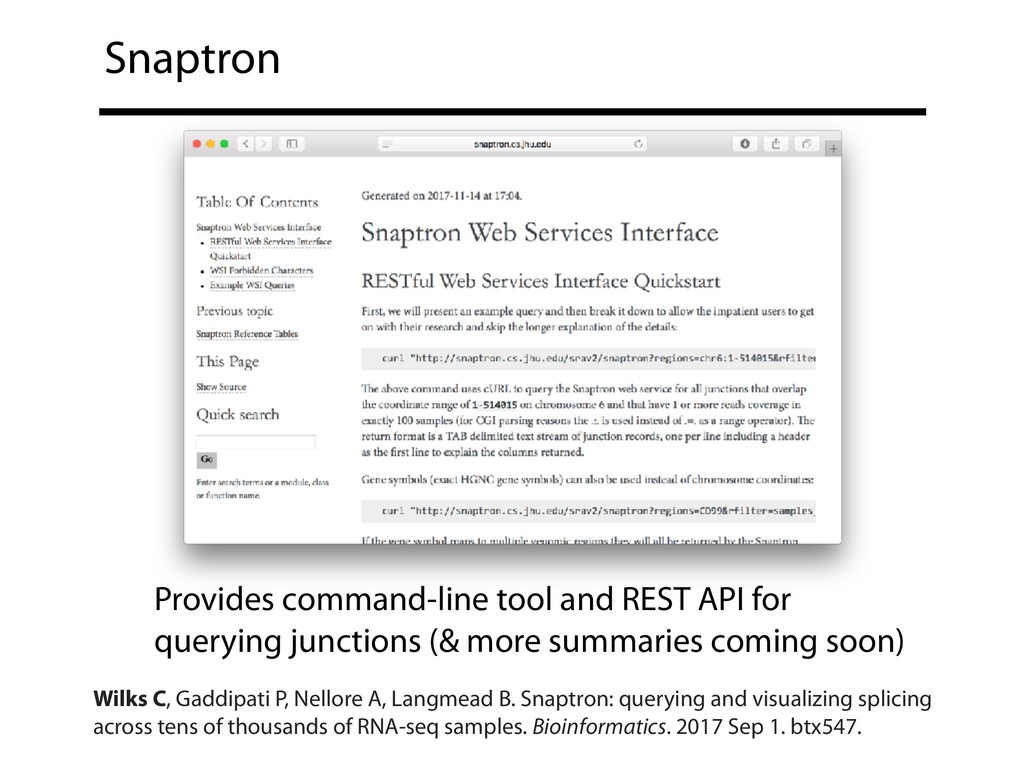
{kind=link}
{kind=link}
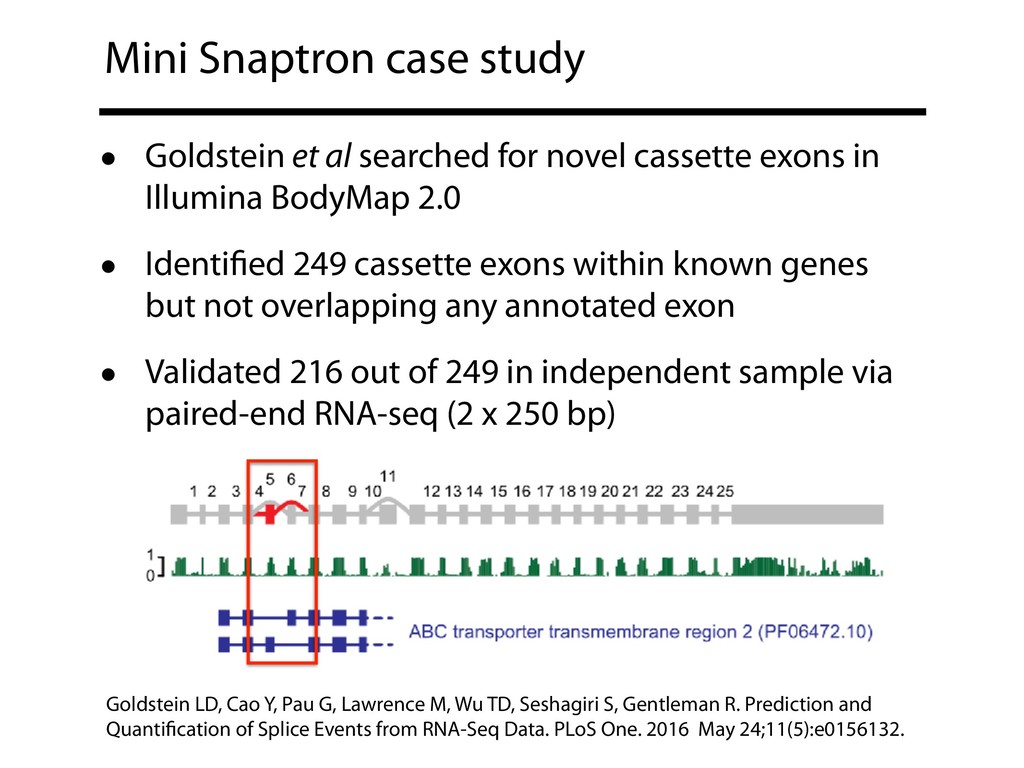
{kind=link}
{kind=link}
{kind=link}
{kind=link}
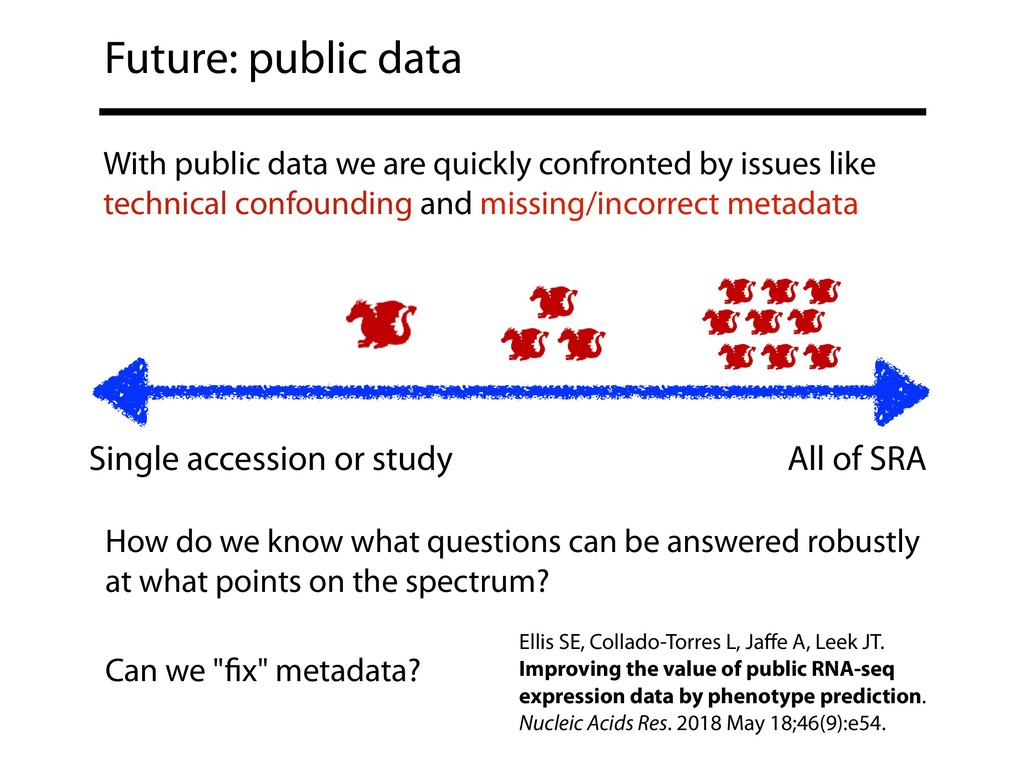
{kind=link}