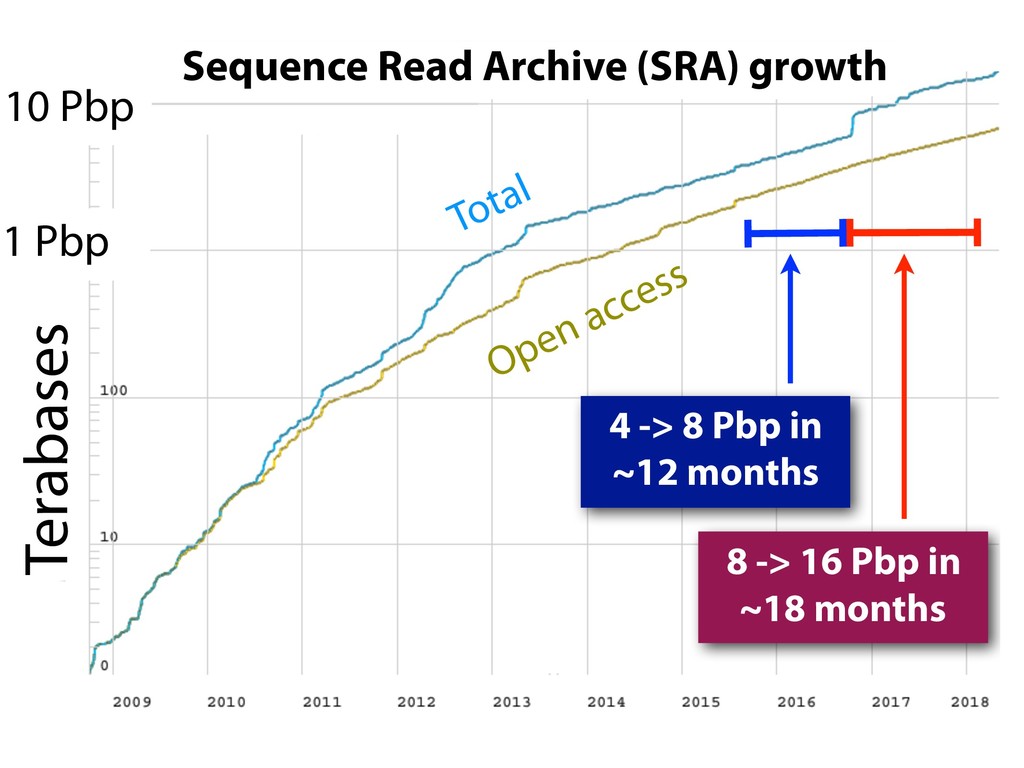
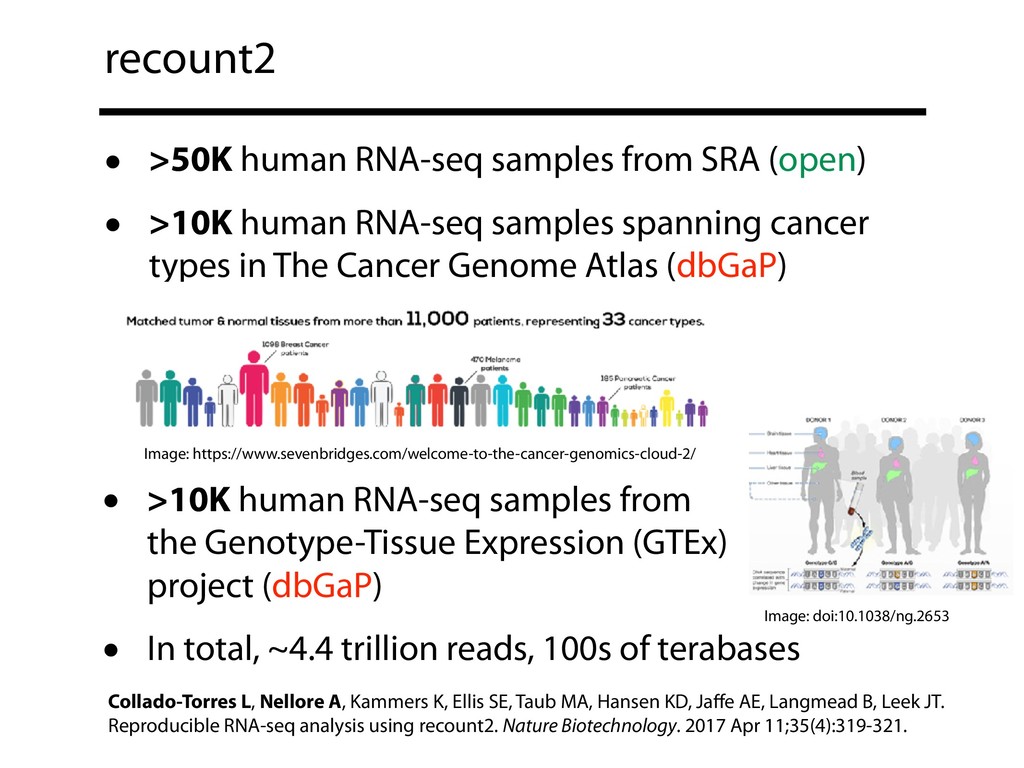
The Sequence Read Archive now contains over a million accessions, including over 200K RNA-seq runs for mouse and over 160K for human. Large-scale projects like GTEx, ICGC and TOPmed are major contributors and huge projects on the horizon, such as the All of Us and Million Veterans programs, will further accelerate this growth. These archives are potential gold mines for researchers but they are not organized for everyday use by scientists. The situation resembles the early days of the World Wide Web, before search engines made the web easy to use.
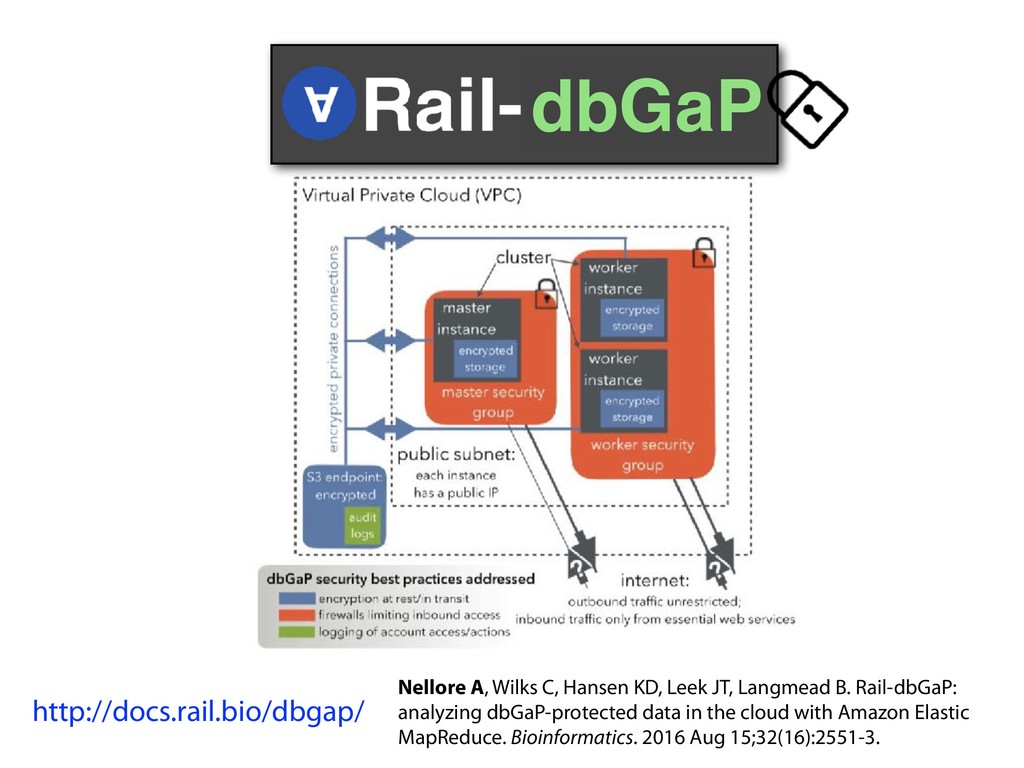
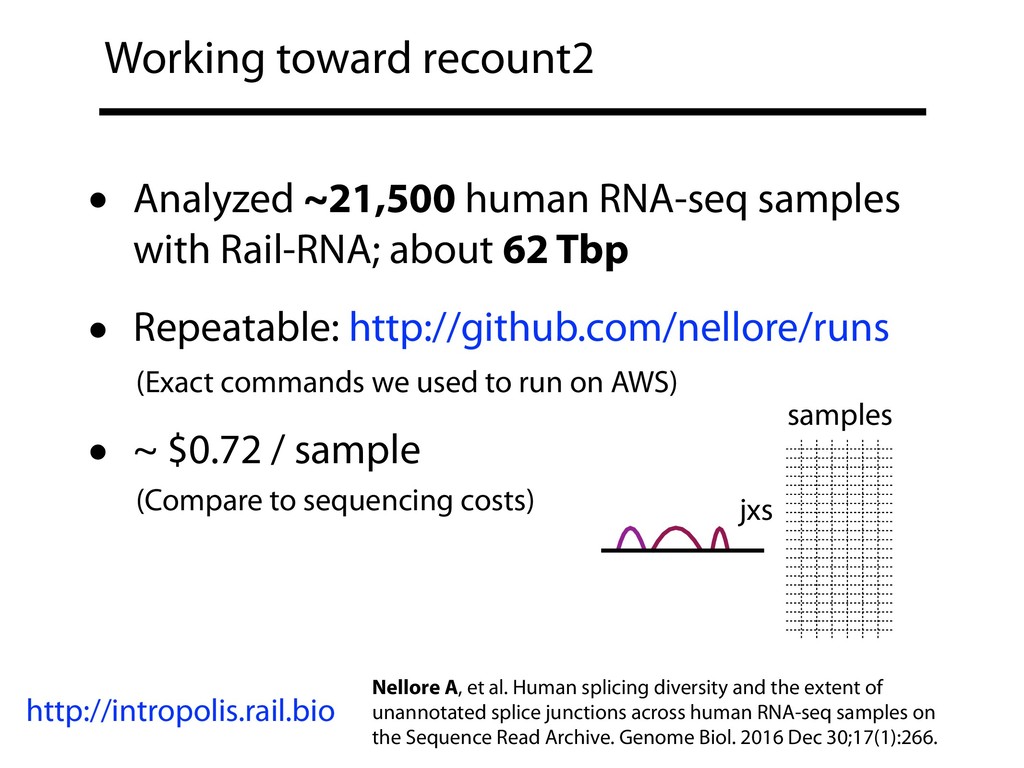
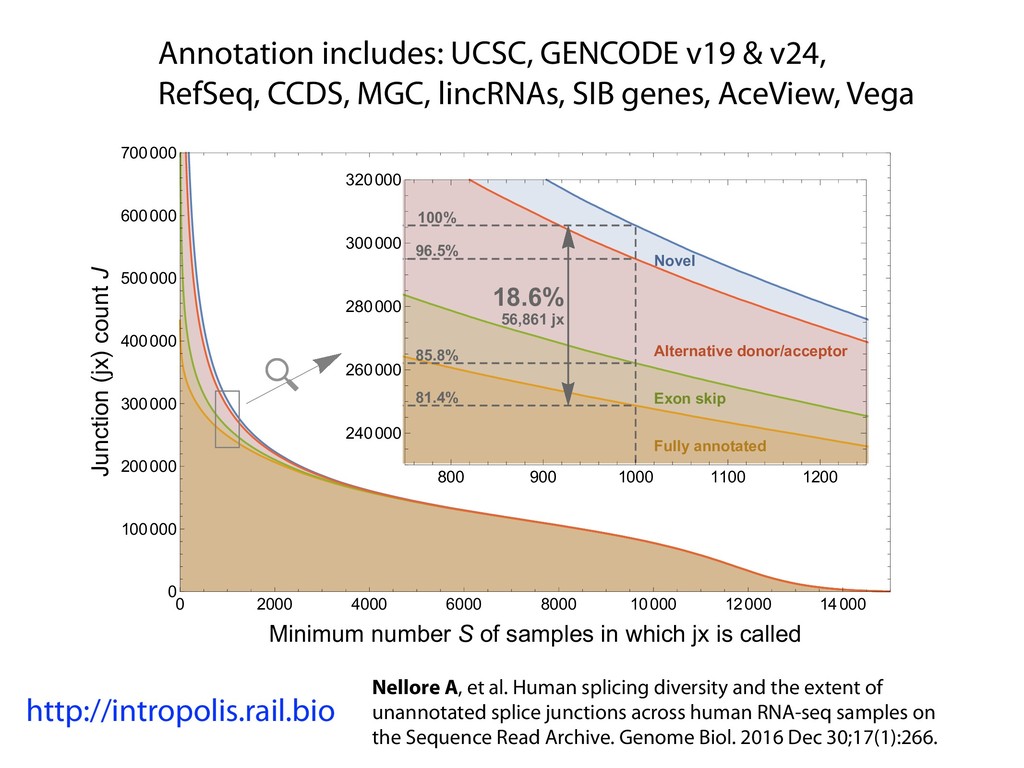
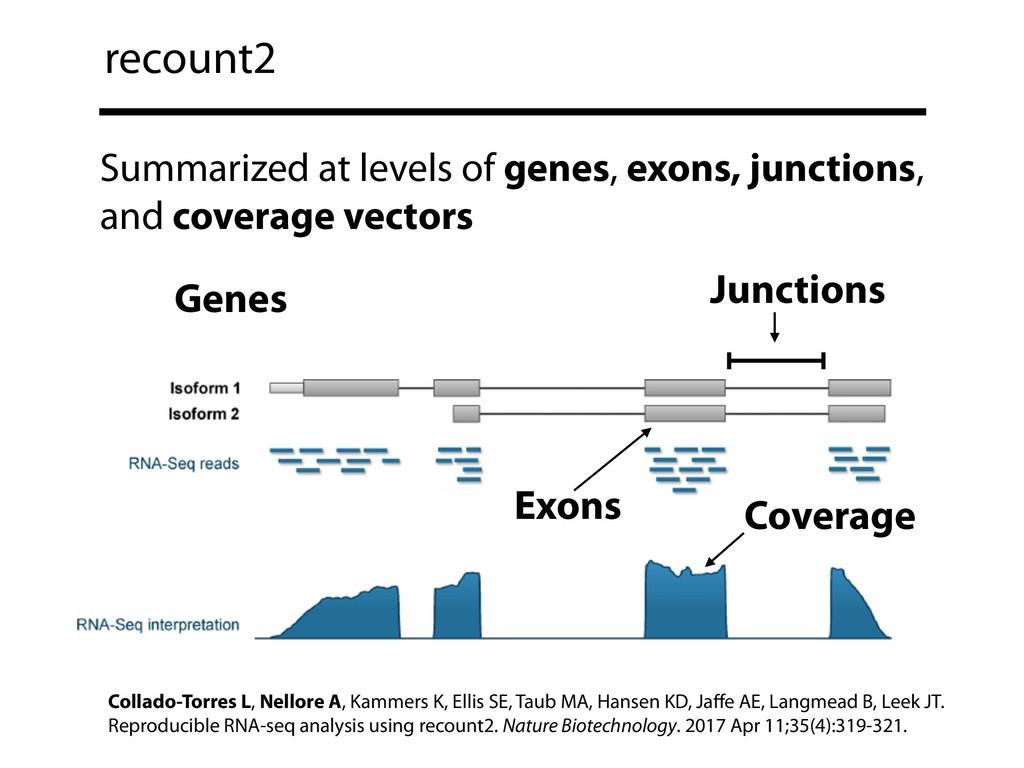
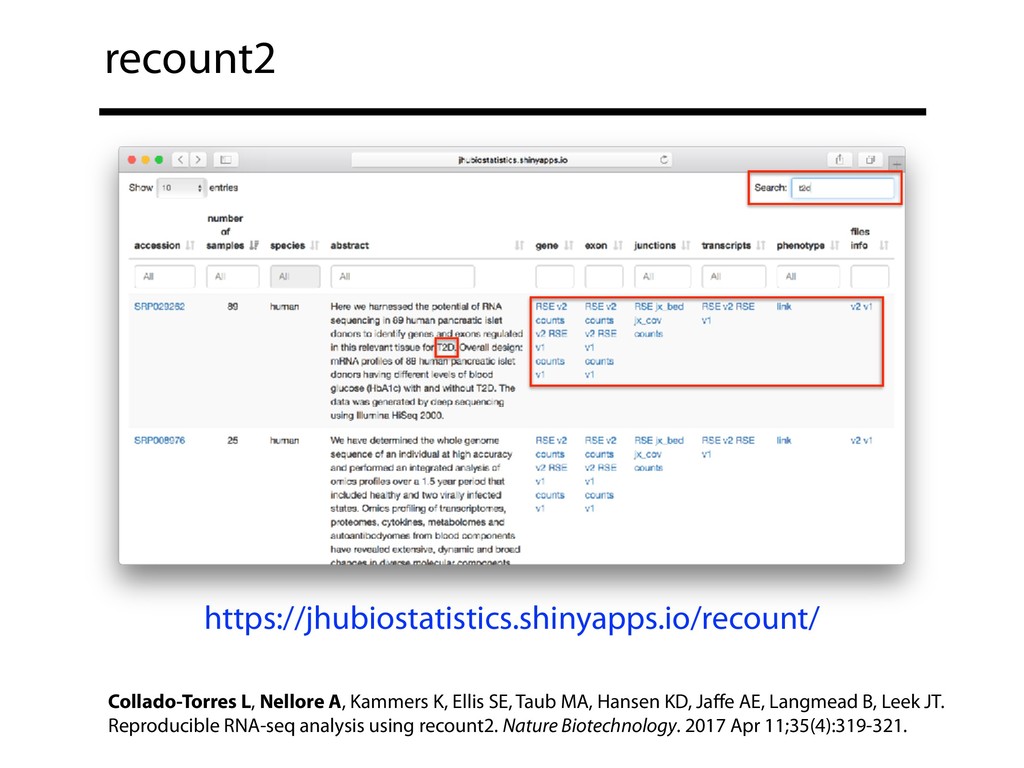
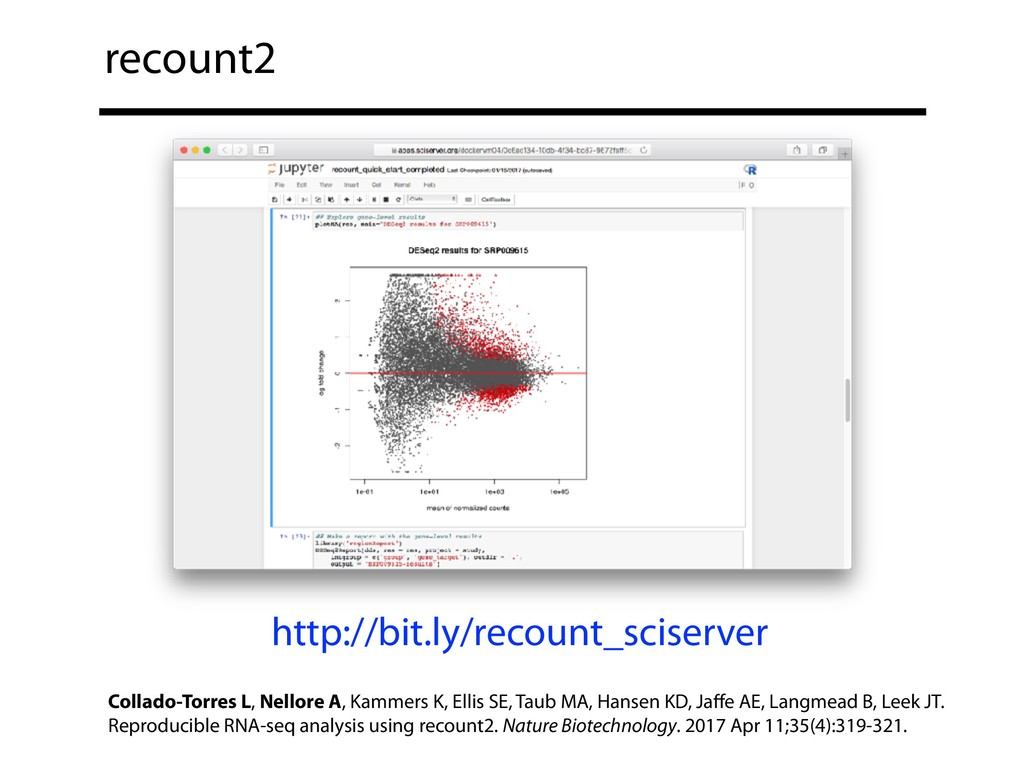
I will describe our progress toward the goal of making it easy for researchers to ask scientific questions about public RNA-seq datasets. I will highlight the Rail/recount2 system for ingesting and summarizing public data and the Snaptron service that makes it queryable. Finally, I will discuss scientific collaborations with neuroscientists and cancer researchers where we applied these tools to perform both targeted queries and large-scale screens. I will highlight ways in which we are learning to make our tools better suited to how scientists work. This is joint work with Abhinav Nellore, Chris Wilks, Jonathan Ling, Luigi Marchionni, Jeff Leek, Kasper Hansen, Andrew Jaffe and others.
![Ben Langmead Assistant Professor, JHU Computer Science [email protected], langmead-lab.org, @BenLangmead](https://files.speakerdeck.com/presentations/7f81f0d11a414891ab8a402da455cdb0/slide_0.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
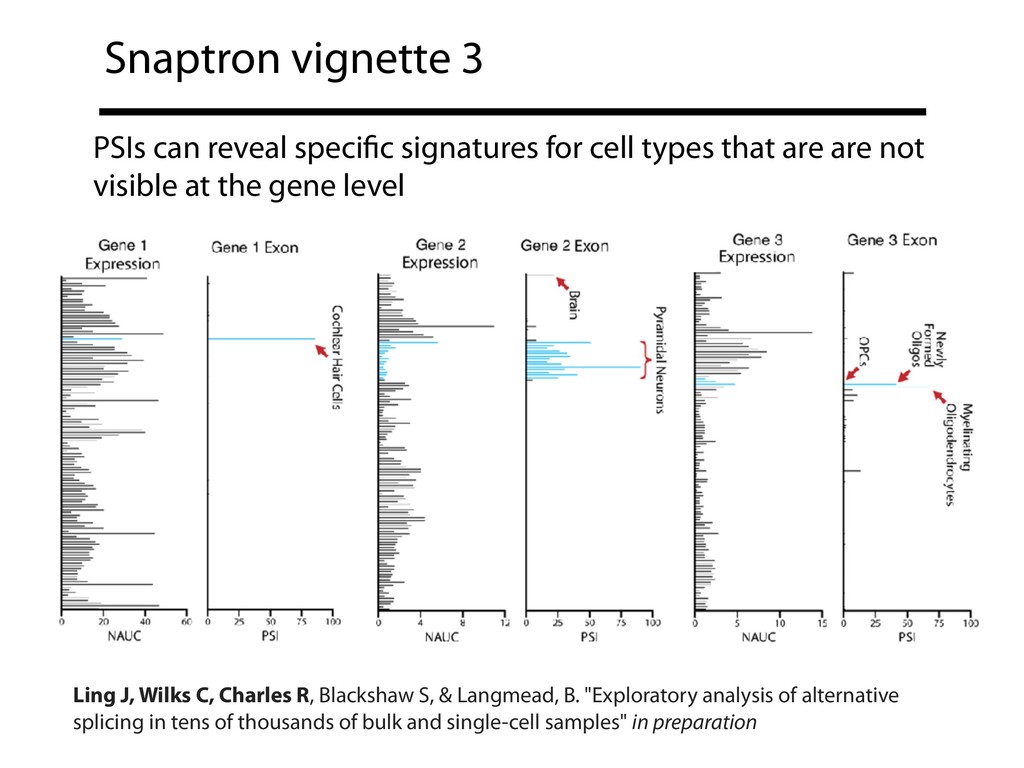{kind=link}
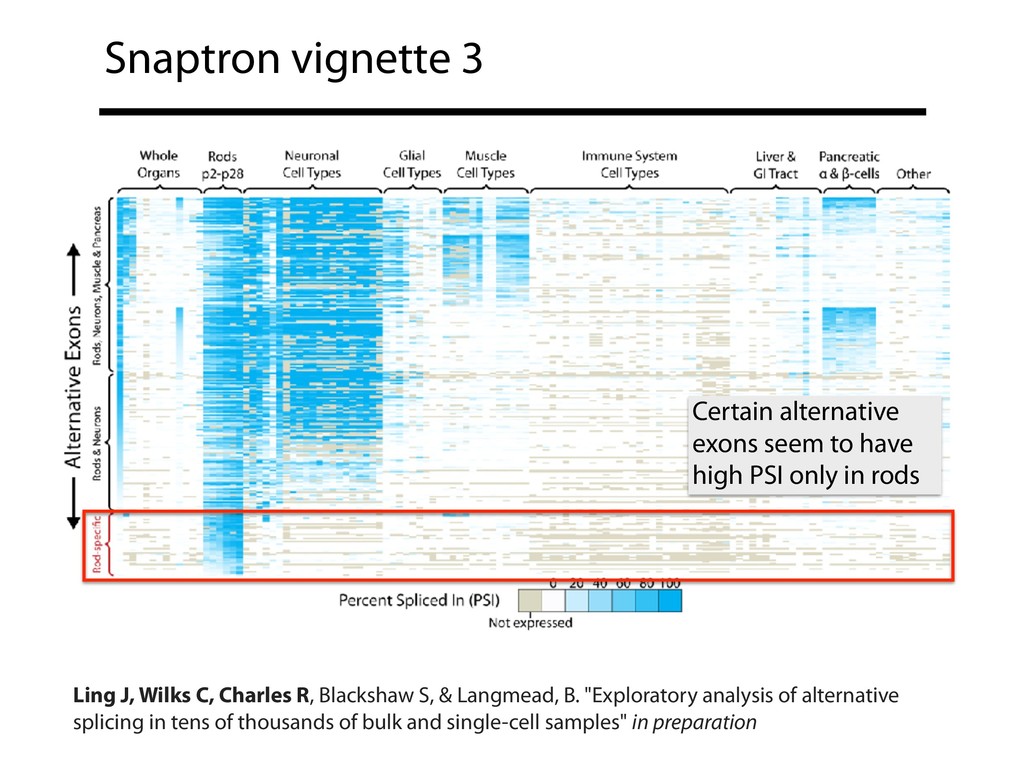{kind=link}
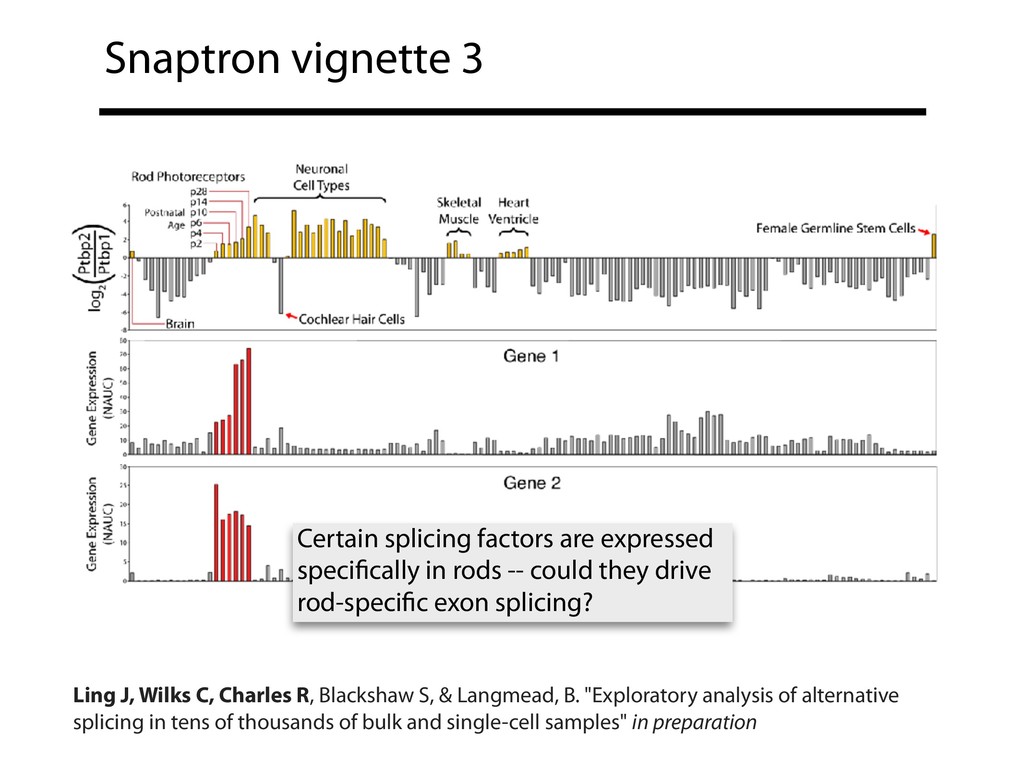{kind=link}
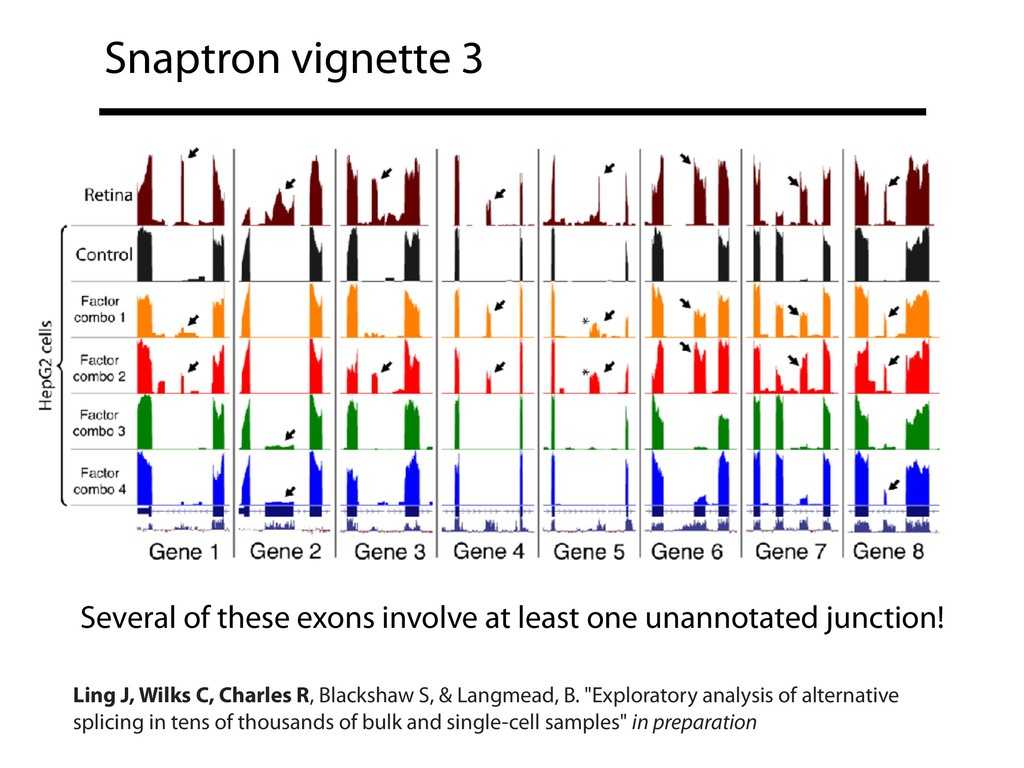{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}