Novak AM, Rosen Y, Haussler D, Paten B. Canonical, stable, general mapping using context schemes. Bioinformatics. 2015 Nov 15;31(22):3569-76. But how do you... Predict mapping quality? Characterize what the heuristics are missing? Select an appropriate scoring scheme? See John Kececioglu's talk!
-6 -5 -3 C -6 2 -6 -6 -5 -3 G -6 -6 2 -6 -5 -3 T -6 -6 -6 2 -5 -3 op -5 -5 -5 -5 ex -3 -3 -3 -3 A C G T op ex A 2 -4 -4 -4 -4 -2 C -4 2 -4 -4 -4 -2 G -4 -4 2 -4 -4 -2 T -4 -4 -4 2 -4 -2 op -4 -4 -4 -4 ex -2 -2 -2 -2 Bowtie 2 minimap 2 Affine-gap scoring A C G T op ex A 9 Qu Qu Qu -40 -6 C Qu 9 Qu Qu -40 -6 G Qu Qu 9 Qu -40 -6 T Qu Qu Qu 9 -40 -6 op -40 -40 -40 -40 ex -6 -6 -6 -6 Novoalign Classic: PAM250
graphs for path queries with applications in genome research." IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB) 11.2 (2014): 375-388. Works around the fact that typical scoring scheme ignores known ALTs REF ALT ALT ALT ALT
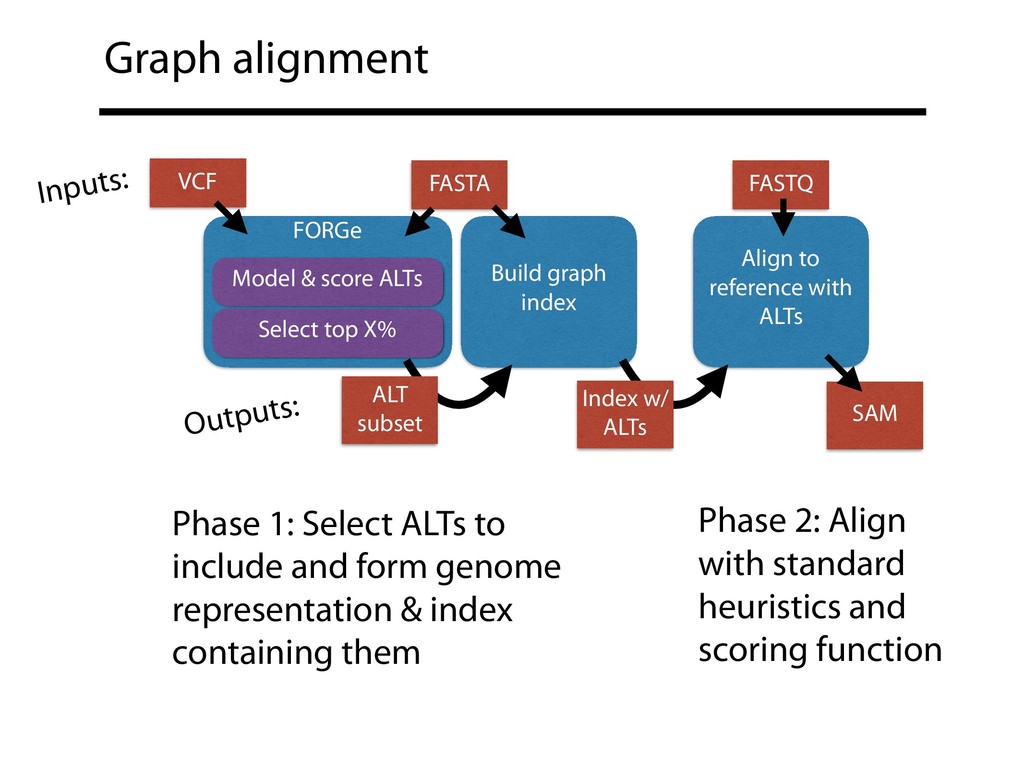
genome representation & index containing them Phase 2: Align with standard heuristics and scoring function FORGe Model & score ALTs Select top X% Build graph index Align to reference with ALTs VCF FASTA FASTQ SAM Inputs: Outputs: ALT subset Index w/ ALTs
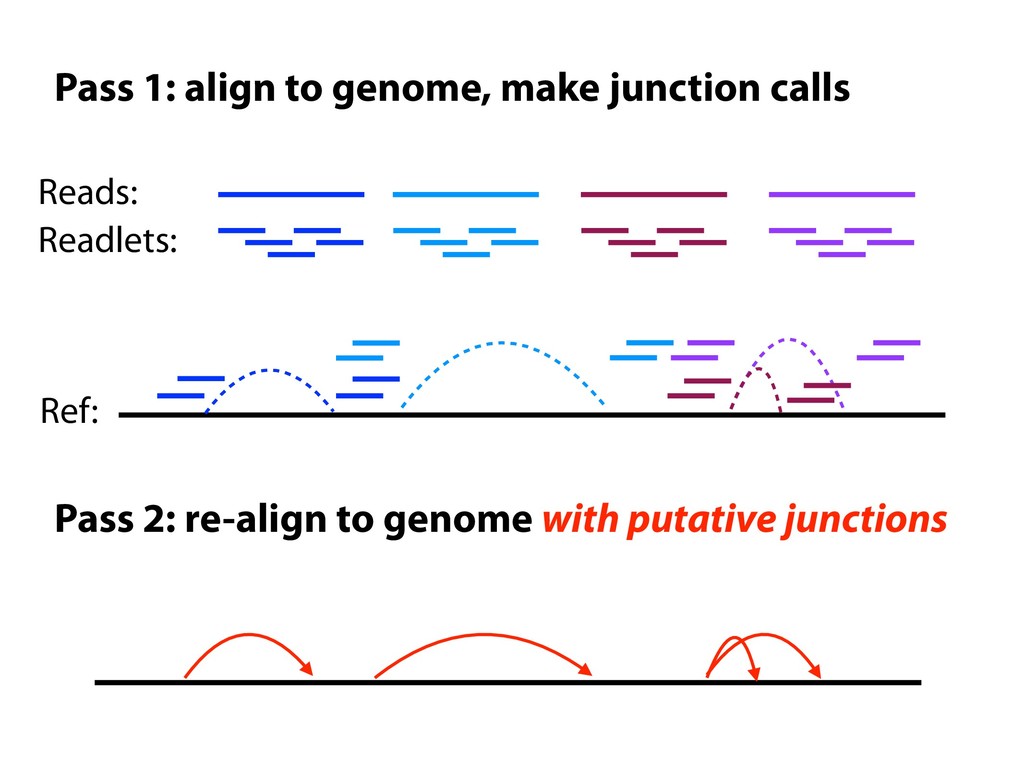
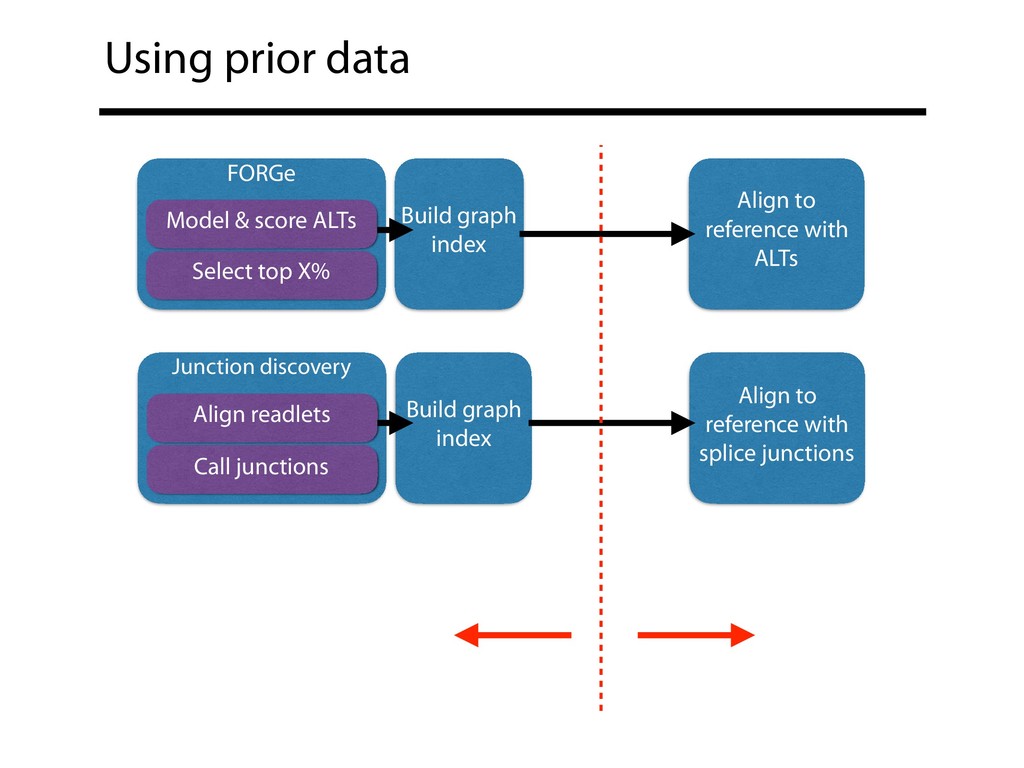
X% Build graph index Align to reference with ALTs Junction discovery Align readlets Call junctions Build graph index Align to reference with splice junctions
alignment -- modify the reference to work around the fact that typical heuristics and scoring functions aren’t appropriate in those settings. Is this a paradigm that we can or should improve on? What can we do differently? Thank you: Jacob Pritt Nae-Chyun Chen NSF: IIS-1349906
![Ben Langmead Assistant Professor, JHU Computer Science [email protected], langmead-lab.org, @BenLangmead](https://files.speakerdeck.com/presentations/f19d9ac0eb0d4d26ae481bdbf8574a02/slide_0.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}