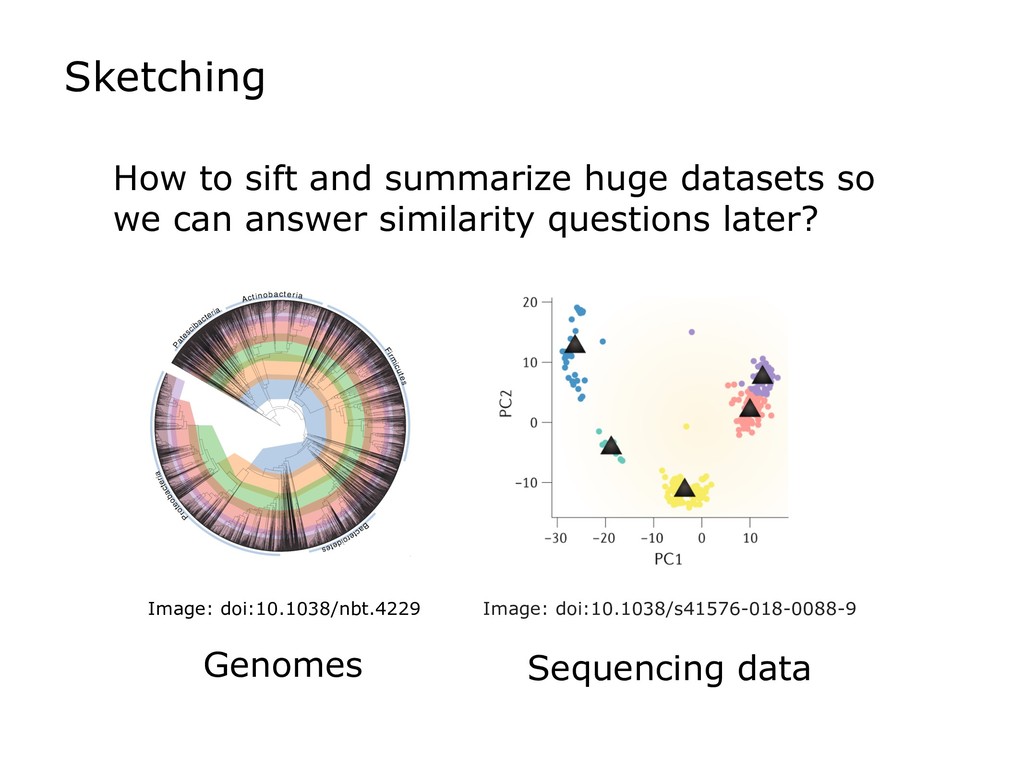
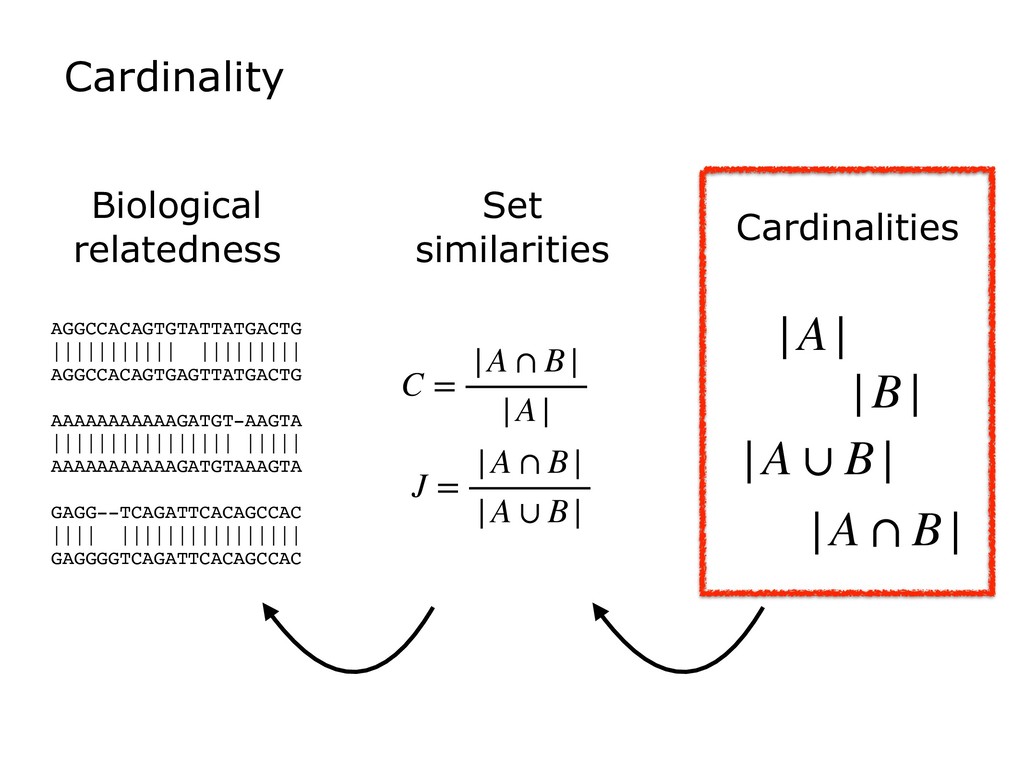
Sketch data structures can be used to distill huge genomics datasets into small summaries. Sketches can then be compared to find, for example, the degree of k-mer similarity between two datasets. This is the basis for a growing number of bionformatics tools solving an array of problems, e.g. clustering genomes, searching for datasets with certain sequence content, accelerating the overlapping step in genome assemblers, or mapping sequencing reads.
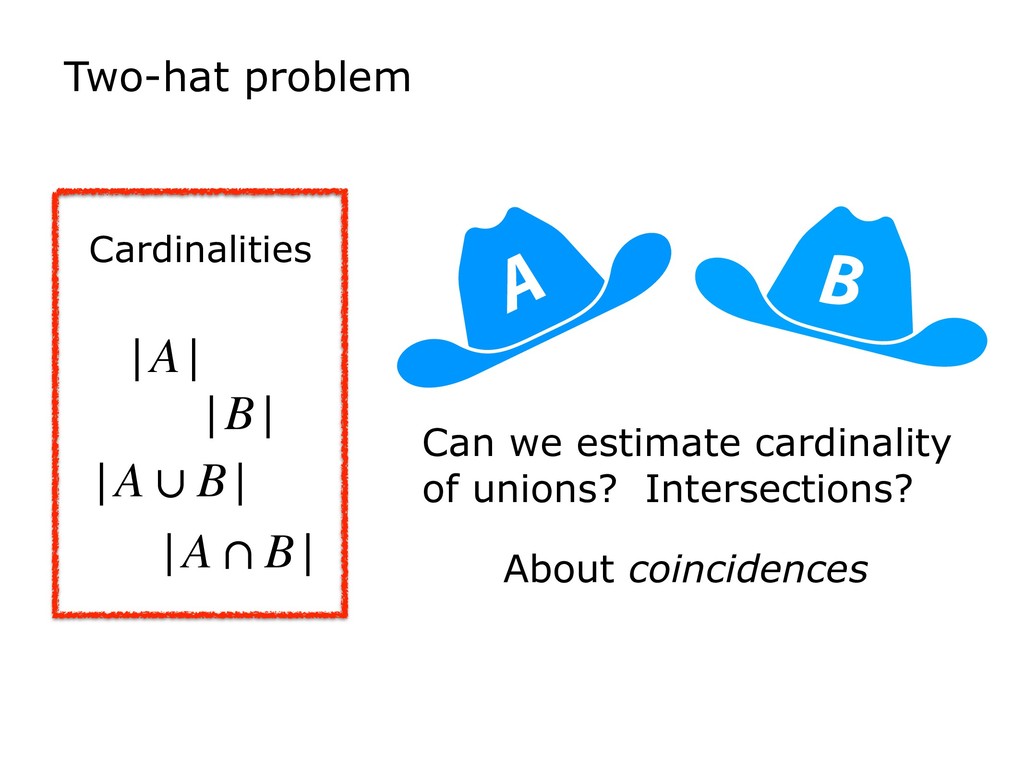
I will discuss the basic problems addressed by sketches, with a focus on MinHash and HyperLogLog. I will suggest a unified way of thinking about these, which are often described in different terms (e.g. "ordered" versus "bit-pattern observable"). I will further relate these structures to Bloom filters, which have found many applications in bioinformatics.
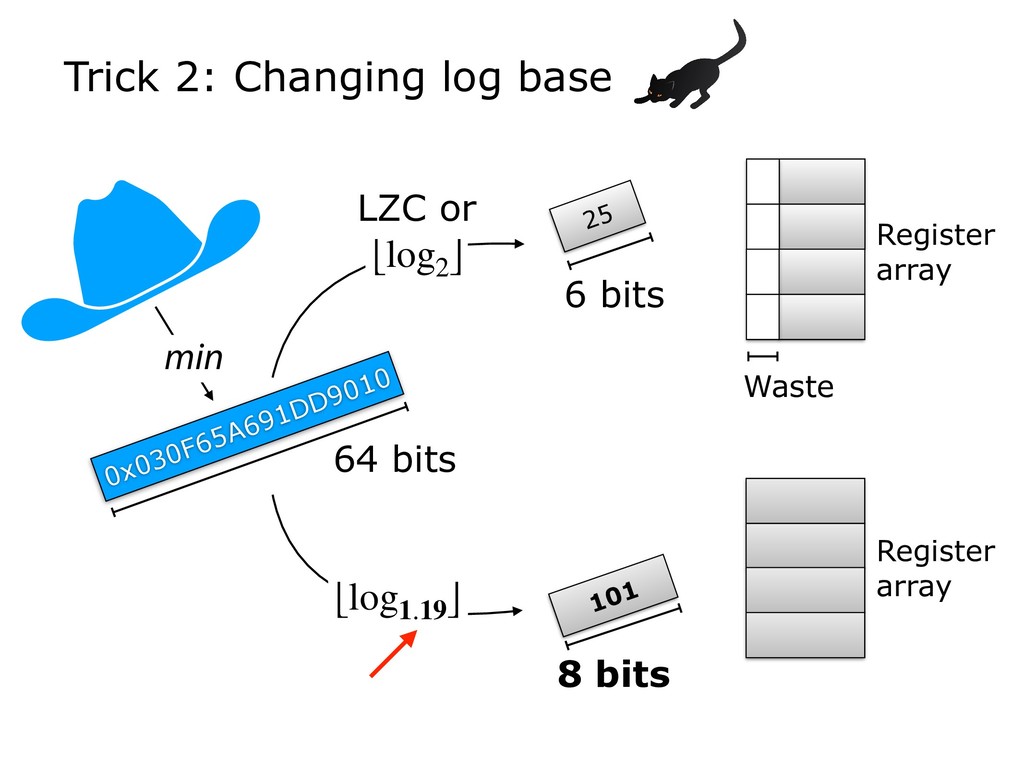
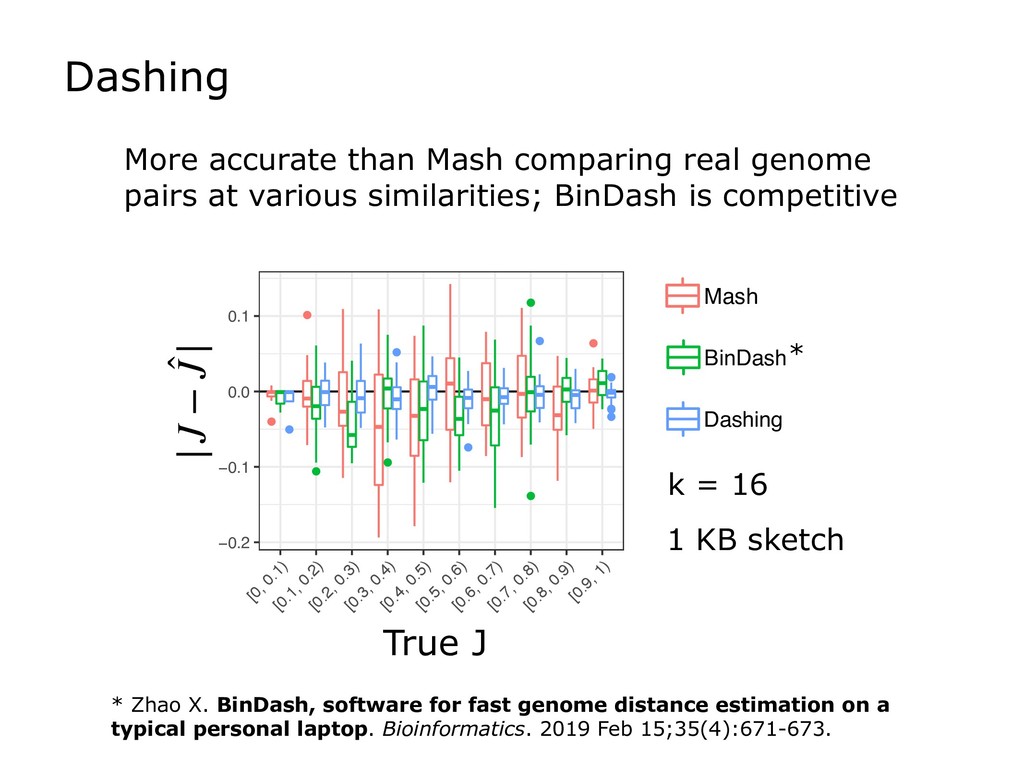
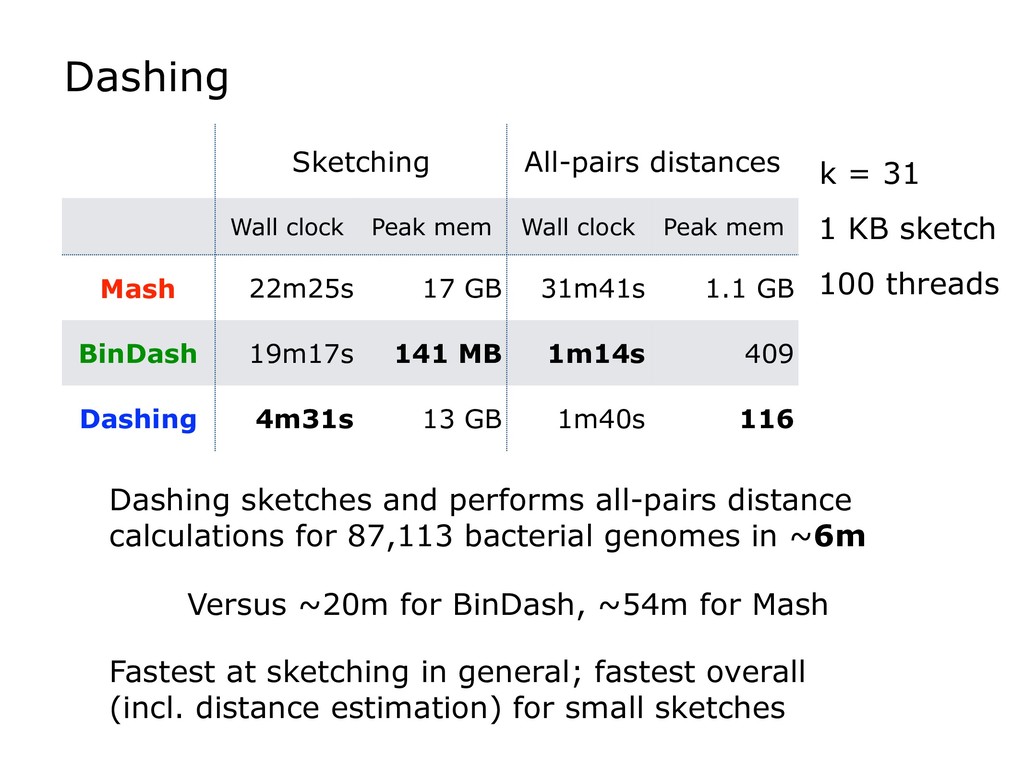
Finally, I will discuss how HyperLogLog is used in the new Dashing software tool, which tackles a similar set of sequence-similarity problems as Mash and BinDash. I will show how HyperLogLog helps address a major issue with MinHash, namely its lower accuracy in cases where one of the sets being compared is much smaller than the other. Finally, I will discuss how the ability to create sketches efficiently enables further accuracy improvements, for example, by making it easier to sketch across an array of k-mer sizes.
![Ben Langmead JHU Computer Science [email protected], langmead-lab.org, @BenLangmead Genome Informatics,](https://files.speakerdeck.com/presentations/c631ca6c3c26466d9ac87d9fc1ee300a/slide_0.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}