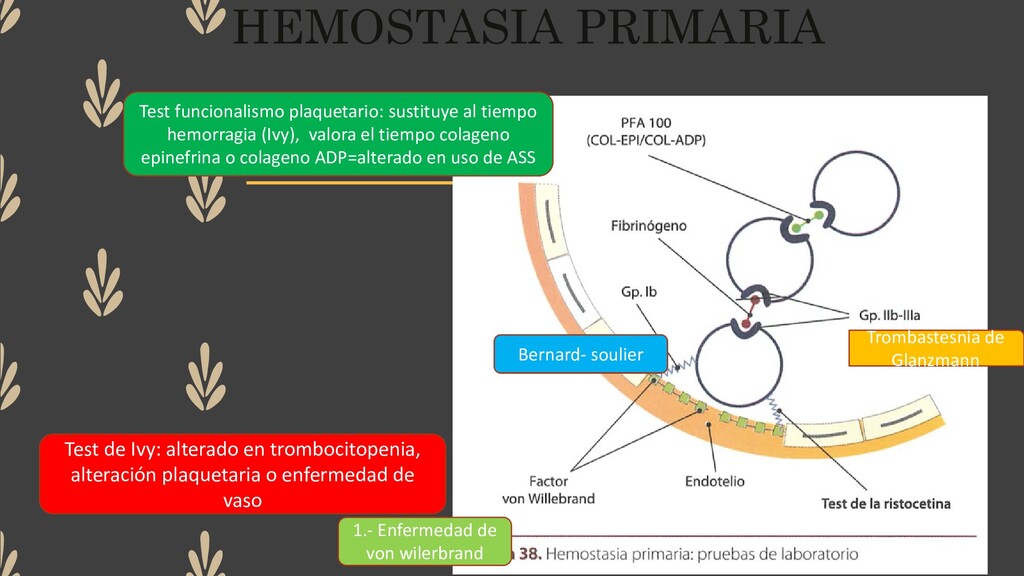
sustituye al tiempo hemorragia (Ivy), valora el tiempo colageno epinefrina o colageno ADP=alterado en uso de ASS Test de Ivy: alterado en trombocitopenia, alteración plaquetaria o enfermedad de vaso 1.- Enfermedad de von wilerbrand
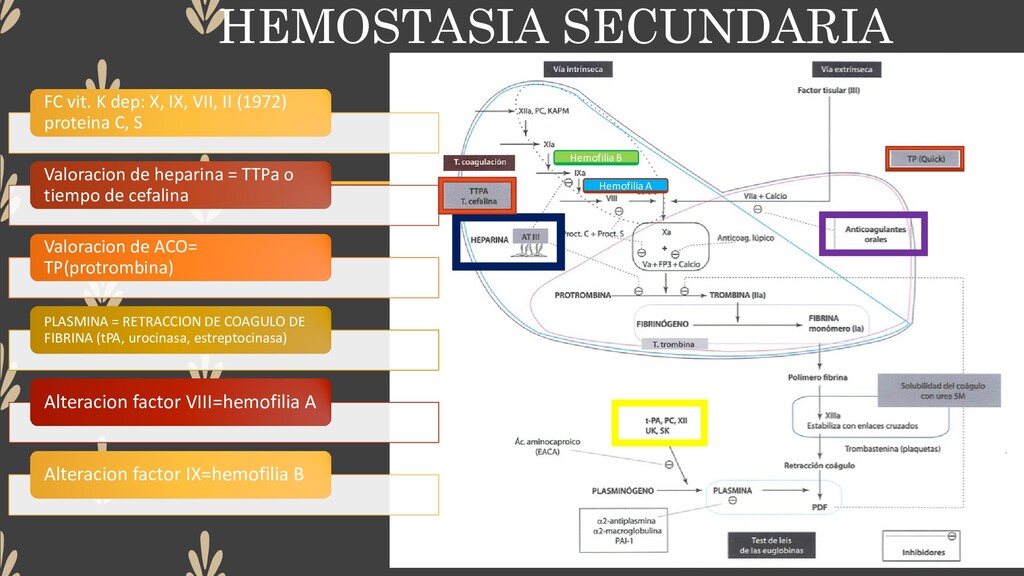
(1972) proteina C, S Valoracion de heparina = TTPa o tiempo de cefalina Valoracion de ACO= TP(protrombina) PLASMINA = RETRACCION DE COAGULO DE FIBRINA (tPA, urocinasa, estreptocinasa) Alteracion factor VIII=hemofilia A Alteracion factor IX=hemofilia B Hemofilia A Hemofilia B
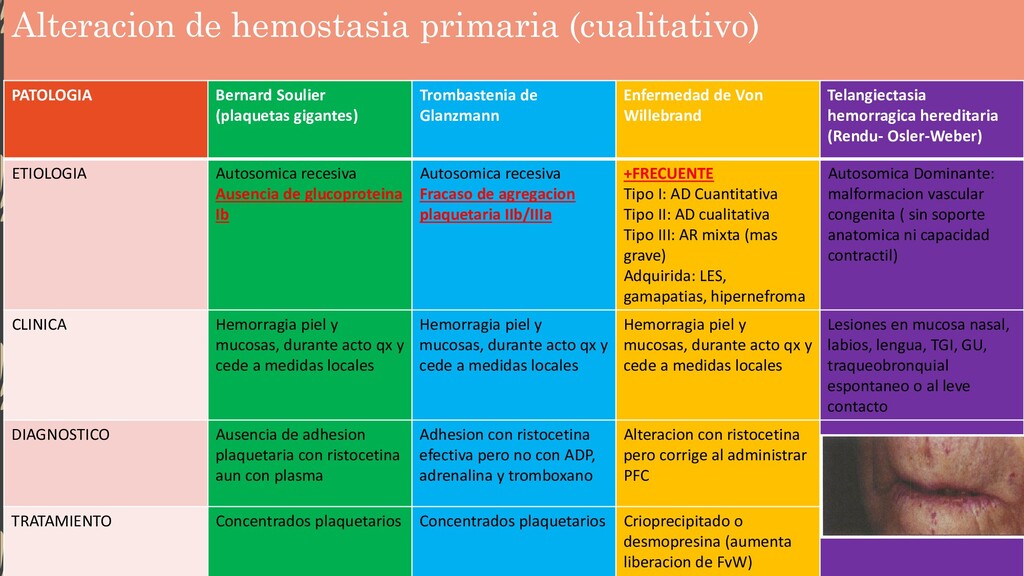
Trombastenia de Glanzmann Enfermedad de Von Willebrand Telangiectasia hemorragica hereditaria (Rendu- Osler-Weber) ETIOLOGIA Autosomica recesiva Ausencia de glucoproteina Ib Autosomica recesiva Fracaso de agregacion plaquetaria IIb/IIIa +FRECUENTE Tipo I: AD Cuantitativa Tipo II: AD cualitativa Tipo III: AR mixta (mas grave) Adquirida: LES, gamapatias, hipernefroma Autosomica Dominante: malformacion vascular congenita ( sin soporte anatomica ni capacidad contractil) CLINICA Hemorragia piel y mucosas, durante acto qx y cede a medidas locales Hemorragia piel y mucosas, durante acto qx y cede a medidas locales Hemorragia piel y mucosas, durante acto qx y cede a medidas locales Lesiones en mucosa nasal, labios, lengua, TGI, GU, traqueobronquial espontaneo o al leve contacto DIAGNOSTICO Ausencia de adhesion plaquetaria con ristocetina aun con plasma Adhesion con ristocetina efectiva pero no con ADP, adrenalina y tromboxano Alteracion con ristocetina pero corrige al administrar PFC TRATAMIENTO Concentrados plaquetarios Concentrados plaquetarios Crioprecipitado o desmopresina (aumenta liberacion de FvW)
(+ FRECUENTE ) Hemofilia B ETIOLOGIA Enfermedad Ligada a X DEFICIENCIA DE FACTOR VIII Enfermedad ligado X DEFICIENCIA DE FACTOR IX CLASIFICACIÓN Leve: 5-40% de actividad Moderada 1-5% de actividad Grave: menor de 1% de actividad Leve: 5-40% de actividad Moderada 1-5% de actividad Grave: menor de 1% de actividad CLÍNICA HEMATOMAS, HEMARTROSIS, HEMORRAGIAS INTERNAS, SANGRADO DESPUES DE LA CIRUGIA HEMATOMAS, HEMARTROSIS, HEMORRAGIAS INTERNAS, SANGRADO DESPUES DE LA CIRUGIA DX TTPa alargado TP normal TTPa alargado TP normal TRATAMIENTO Factor VIII recombinante Tx cronico puede generar anticuerpos anti el factor especifico Factor IX recombinante Tx cronico genera anticuerpos anti el factor dado
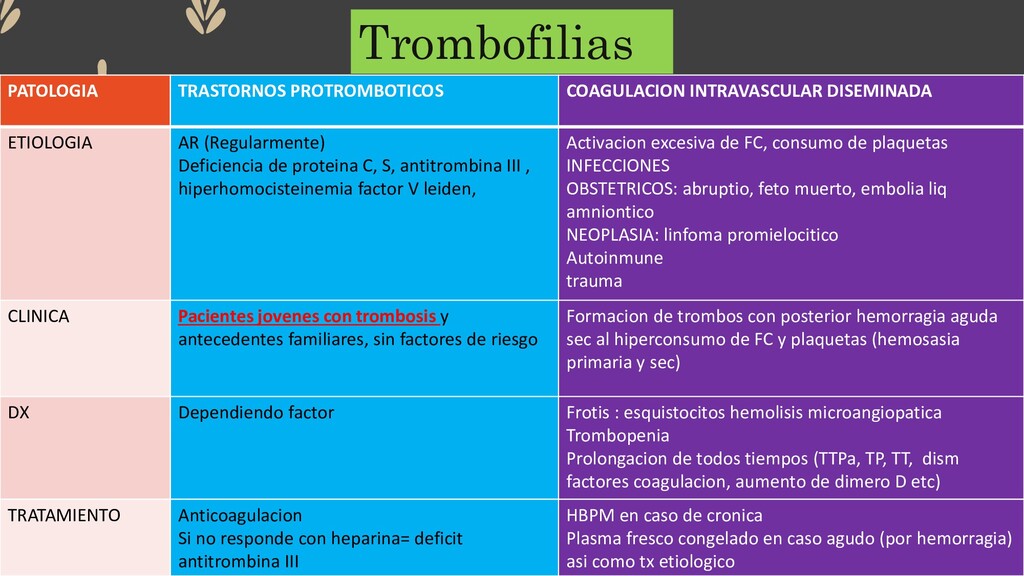
Deficiencia de proteina C, S, antitrombina III , hiperhomocisteinemia factor V leiden, Activacion excesiva de FC, consumo de plaquetas INFECCIONES OBSTETRICOS: abruptio, feto muerto, embolia liq amniontico NEOPLASIA: linfoma promielocitico Autoinmune trauma CLINICA Pacientes jovenes con trombosis y antecedentes familiares, sin factores de riesgo Formacion de trombos con posterior hemorragia aguda sec al hiperconsumo de FC y plaquetas (hemosasia primaria y sec) DX Dependiendo factor Frotis : esquistocitos hemolisis microangiopatica Trombopenia Prolongacion de todos tiempos (TTPa, TP, TT, dism factores coagulacion, aumento de dimero D etc) TRATAMIENTO Anticoagulacion Si no responde con heparina= deficit antitrombina III HBPM en caso de cronica Plasma fresco congelado en caso agudo (por hemorragia) asi como tx etiologico
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}