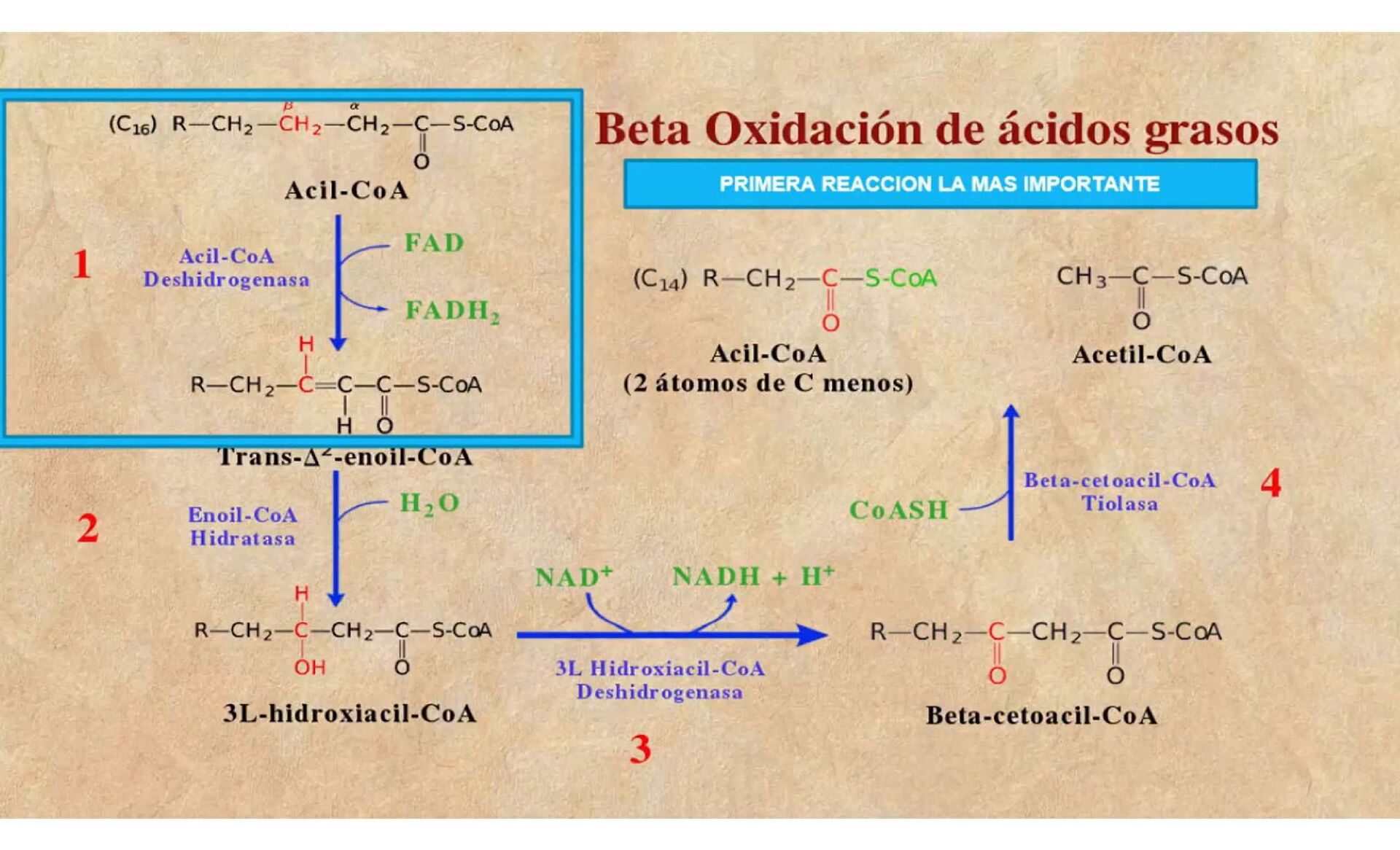
de oxidación de ácidos grasos (DOAG) son un grupo heterogéneo de enfermedades causadas por la deficiencia de alguna enzima que participa en la vía del metabolismo de los ácidos grasos, las enzimas que se han asociado a enfermedad en el humano.
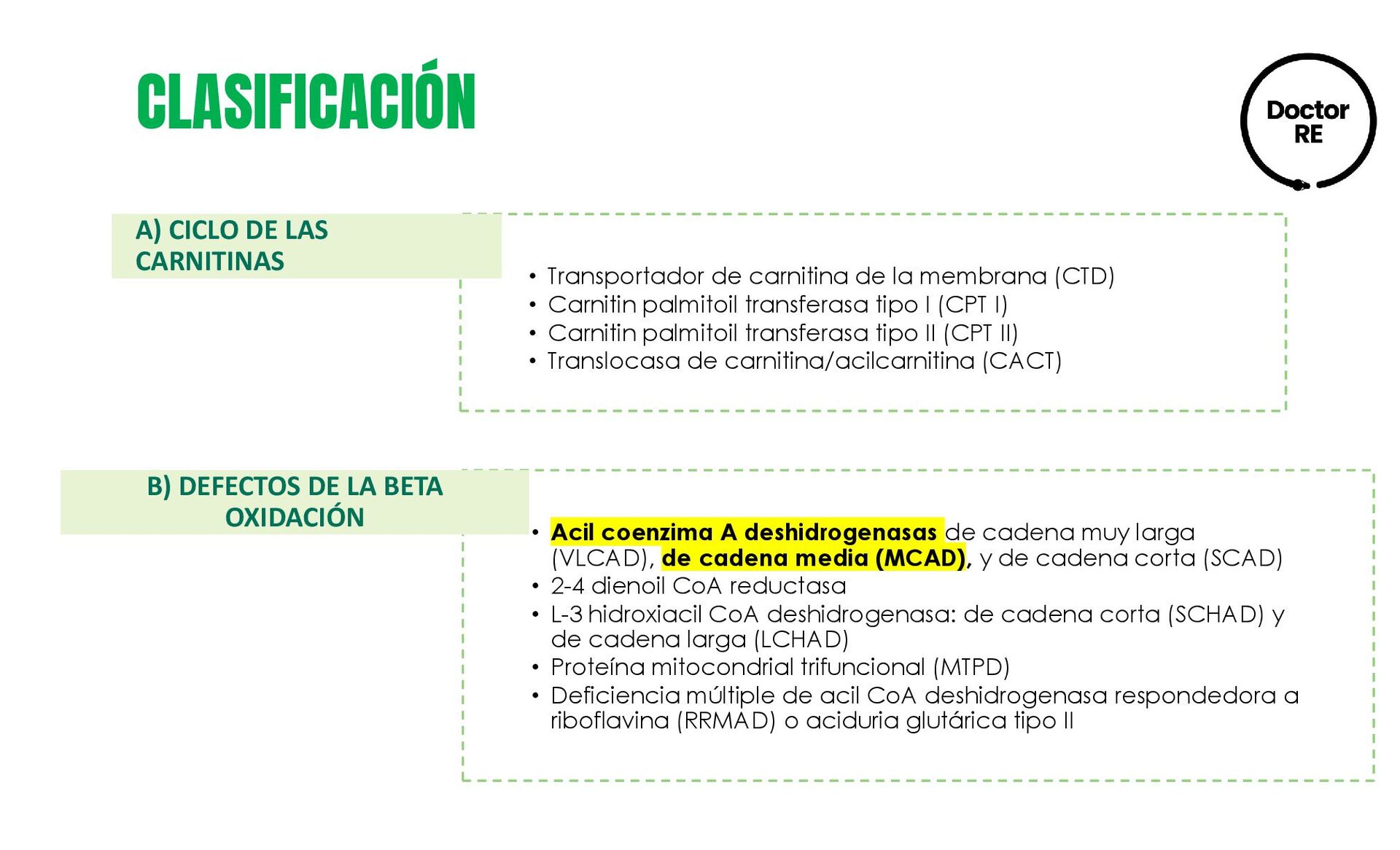
Carnitin palmitoil transferasa tipo I (CPT I) • Carnitin palmitoil transferasa tipo II (CPT II) • Translocasa de carnitina/acilcarnitina (CACT) • Acil coenzima A deshidrogenasas de cadena muy larga (VLCAD), de cadena media (MCAD), y de cadena corta (SCAD) • 2-4 dienoil CoA reductasa • L-3 hidroxiacil CoA deshidrogenasa: de cadena corta (SCHAD) y de cadena larga (LCHAD) • Proteína mitocondrial trifuncional (MTPD) • Deficiencia múltiple de acil CoA deshidrogenasa respondedora a riboflavina (RRMAD) o aciduria glutárica tipo II A) CICLO DE LAS CARNITINAS B) DEFECTOS DE LA BETA OXIDACIÓN
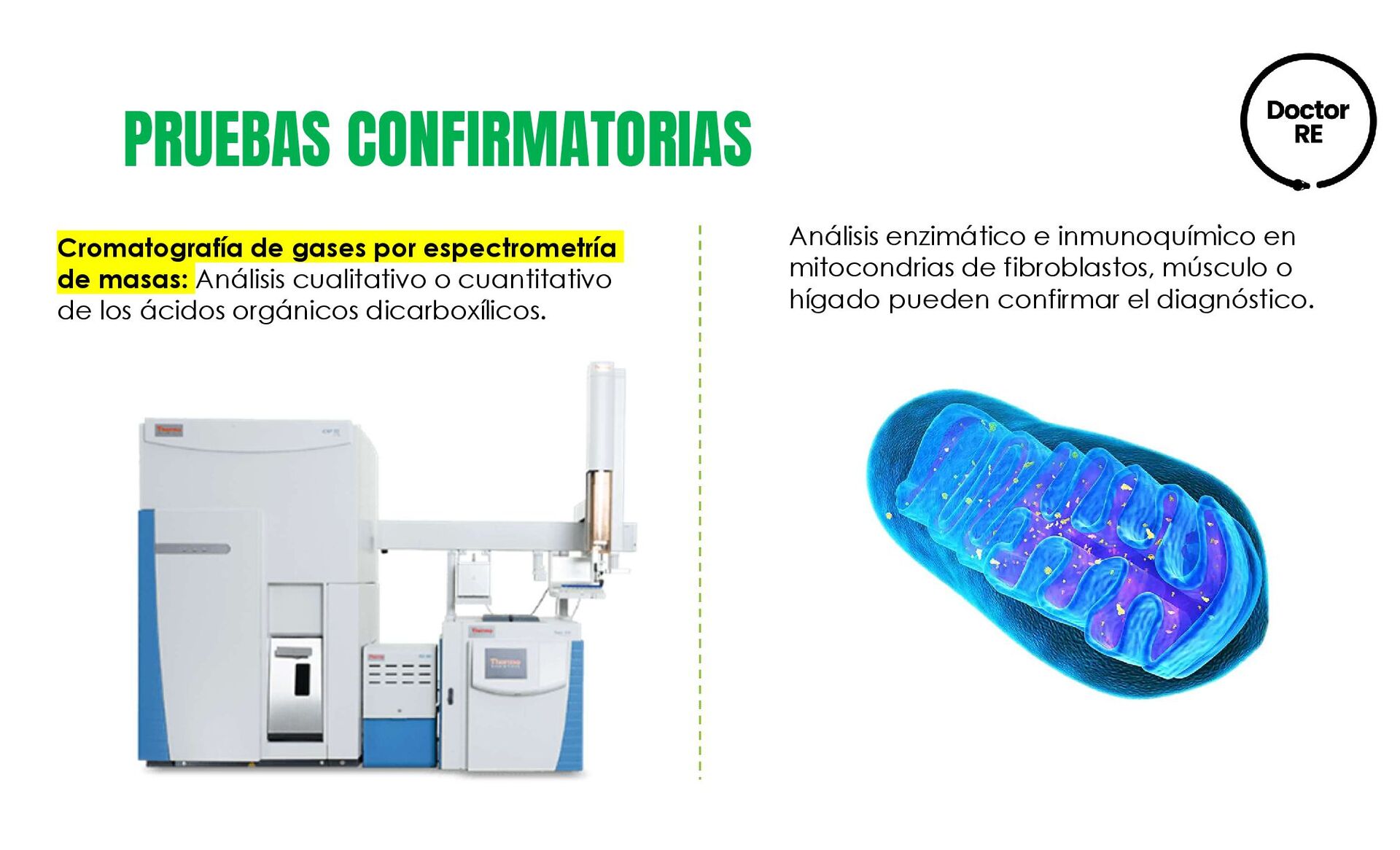
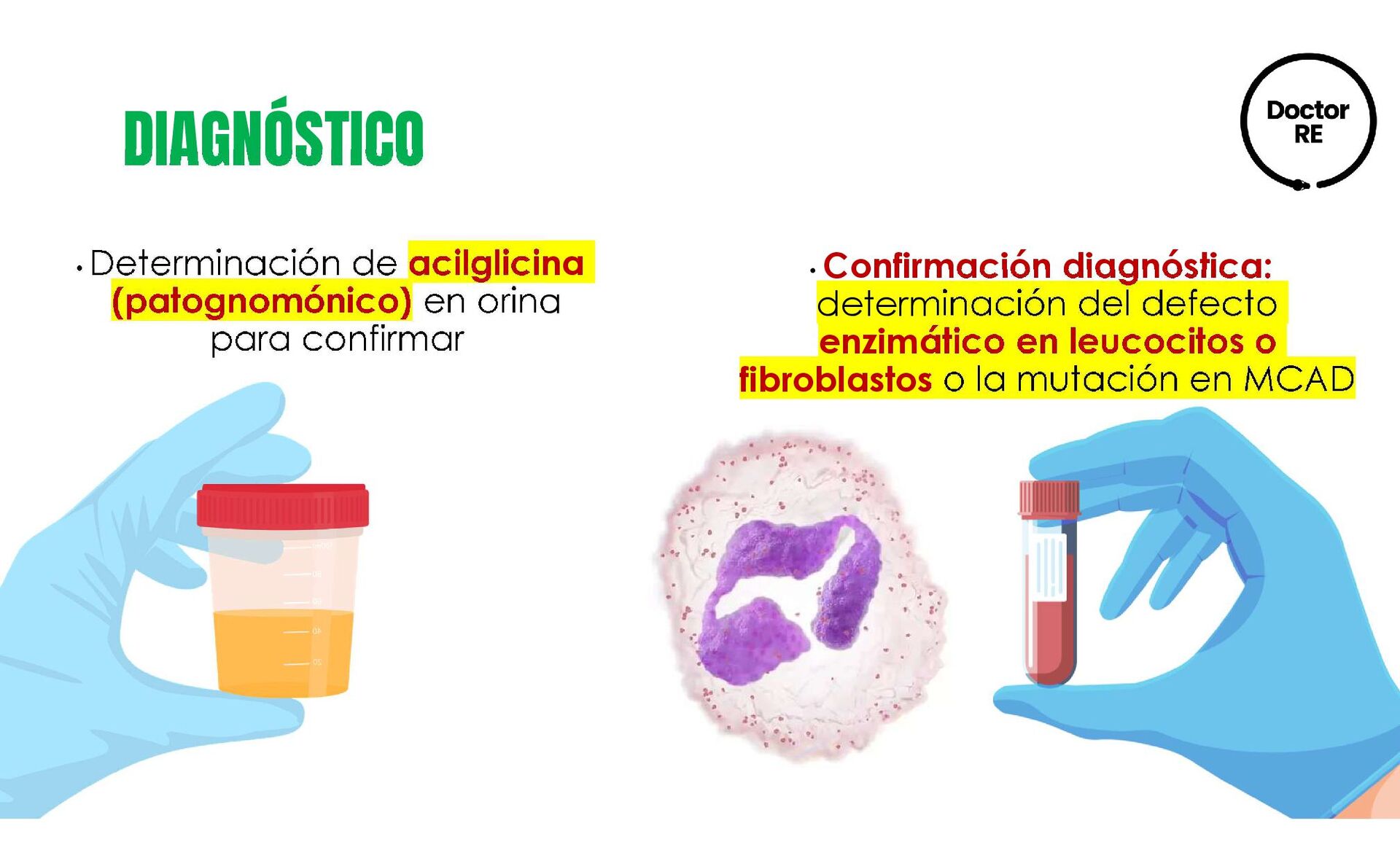
cualitativo o cuantitativo de los ácidos orgánicos dicarboxílicos. Análisis enzimático e inmunoquímico en mitocondrias de fibroblastos, músculo o hígado pueden confirmar el diagnóstico.
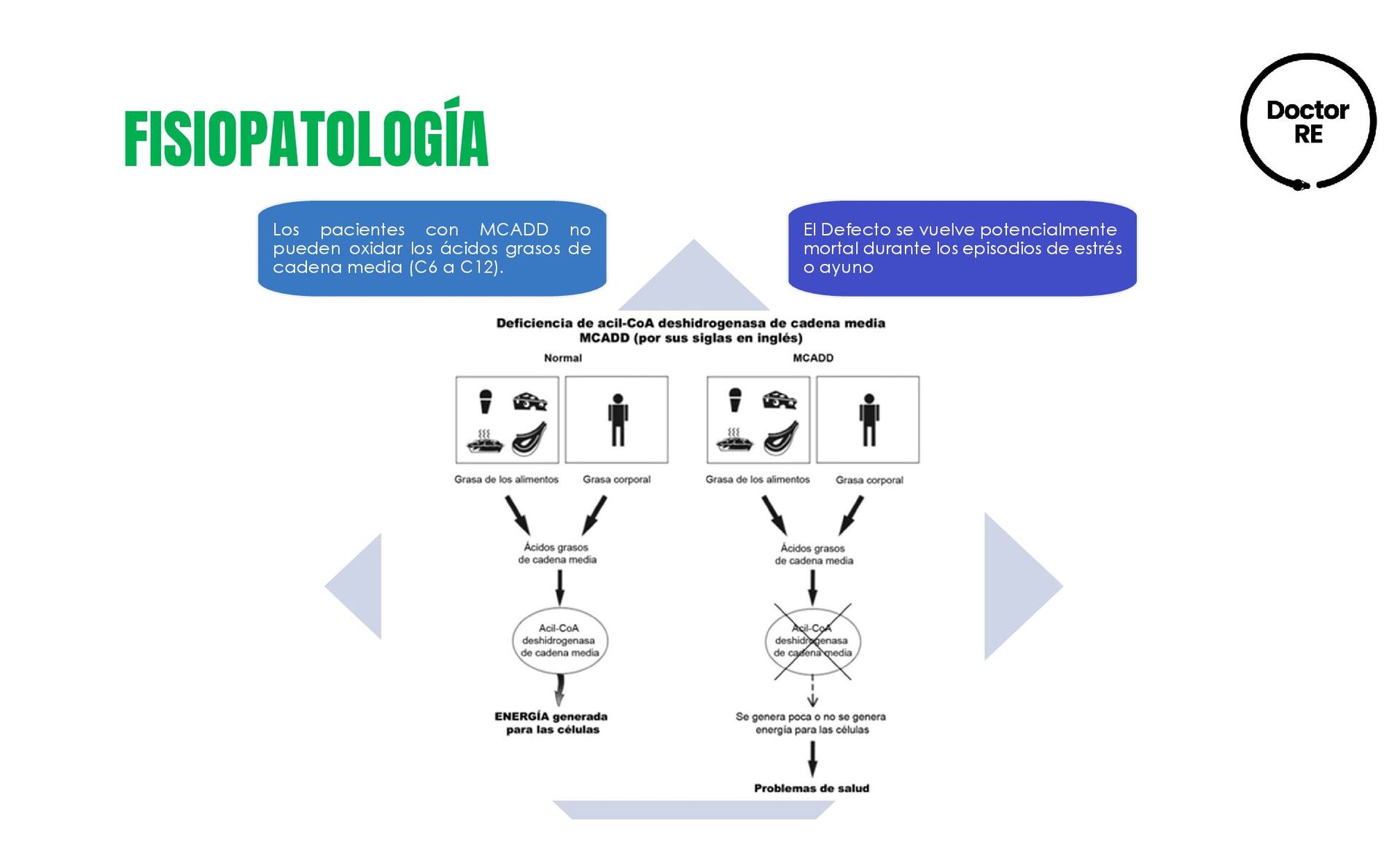
MCADD se produce por el gen ACADM (cromosoma 1p31). Es el defecto de la oxidación mitocondrial de ácidos grasos más común. Se detecta por tamiz neonatal • Tasa de mortalidad 25% durante 1ra manifestación clínica. • La sintomatología puede iniciar entre los 3 meses y 3 años de edad • 3% de los niños que presentan síndrome de muerte súbita del lactante está relacionada con un defecto de la oxidación de ácidos grasos. Frecuencia 1;9,000 a 1:60,000 RNV y se transmite por herencia autosómica recesiva.
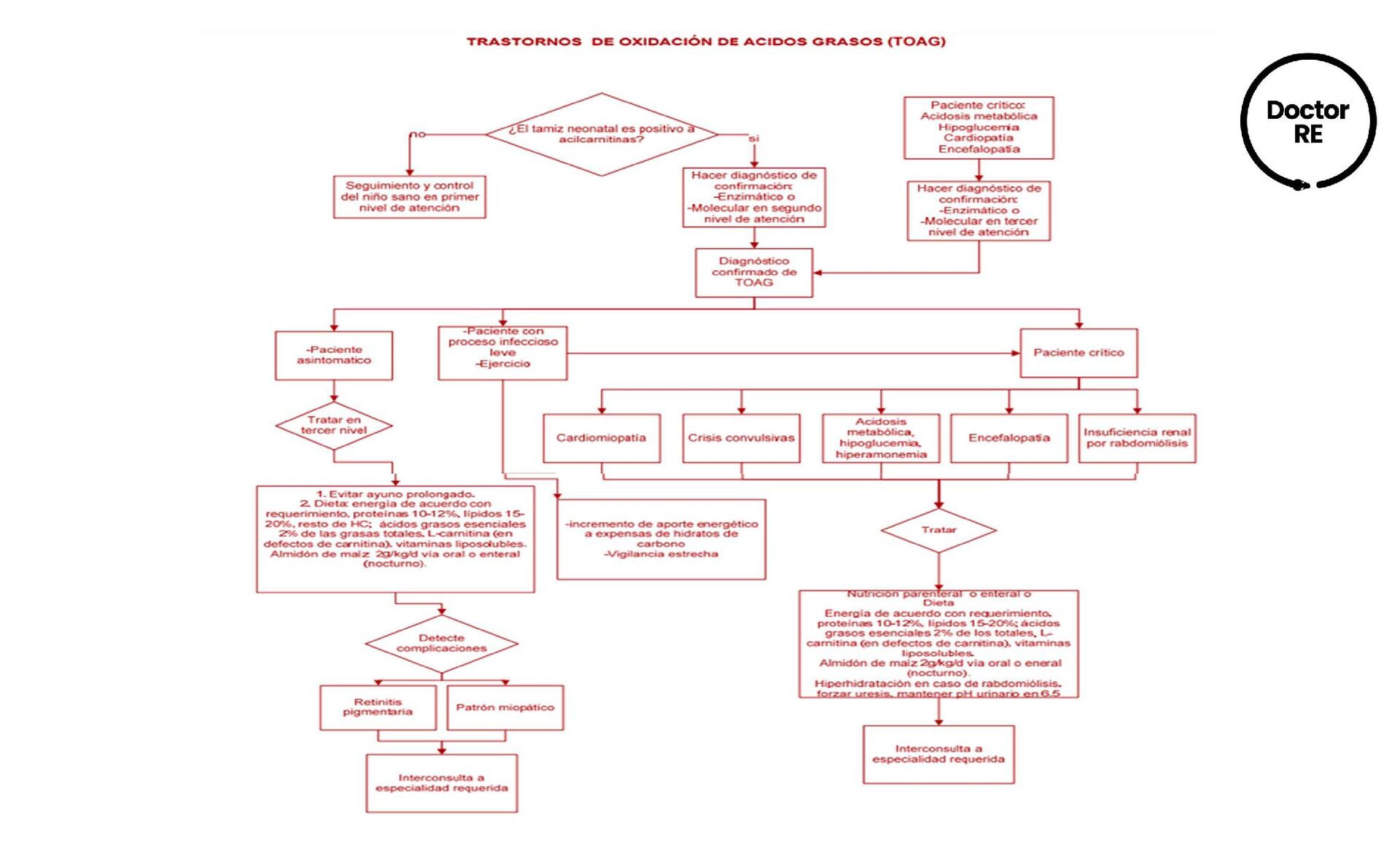
la lipólisis del tejido adiposo, siendo fundamental en el periodo neonatal y en descompensación metabólica Fase aguda: se debe de intervenir con nutrición parenteral, con glucosa intravenosa en dosis de 10 mg/kg/min ó más para mantener glucemia en 100 mg/dl. Fase crónica: el objetivo principal es prevenir la hipoglucemia a través de una dieta fraccionada para evitar periodos de ayuno. En niños menores el ayuno no debe superar las 4 hrs y en mayores 8 hrs Vigilancia clínica es muy importante y los principales objetivos son la vigilancia de crecimiento, desarrollo y la dieta. Estudios bioquímicos para el seguimiento y control incluyen: la determinación de carnitina libre y acilcarnitinas
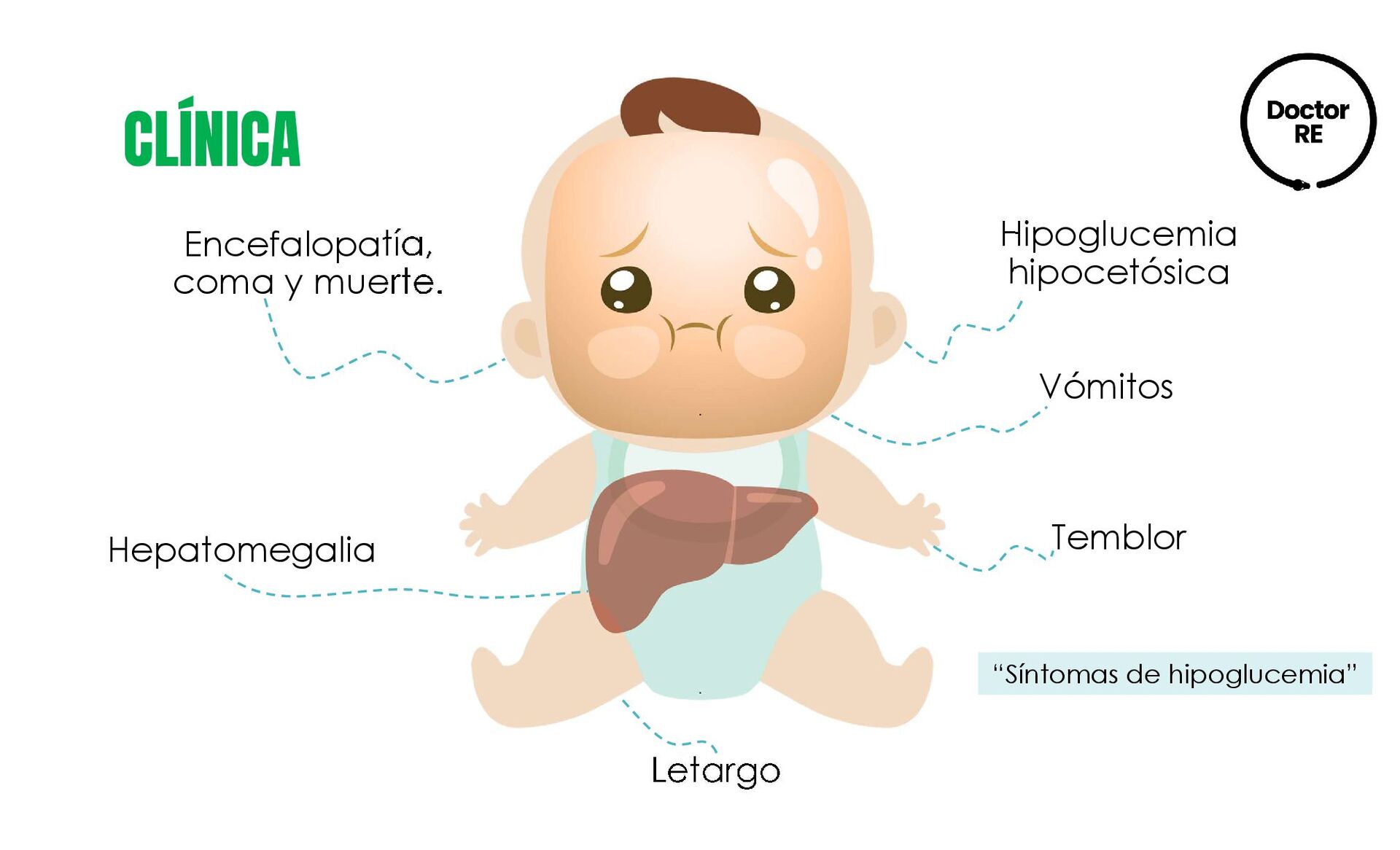
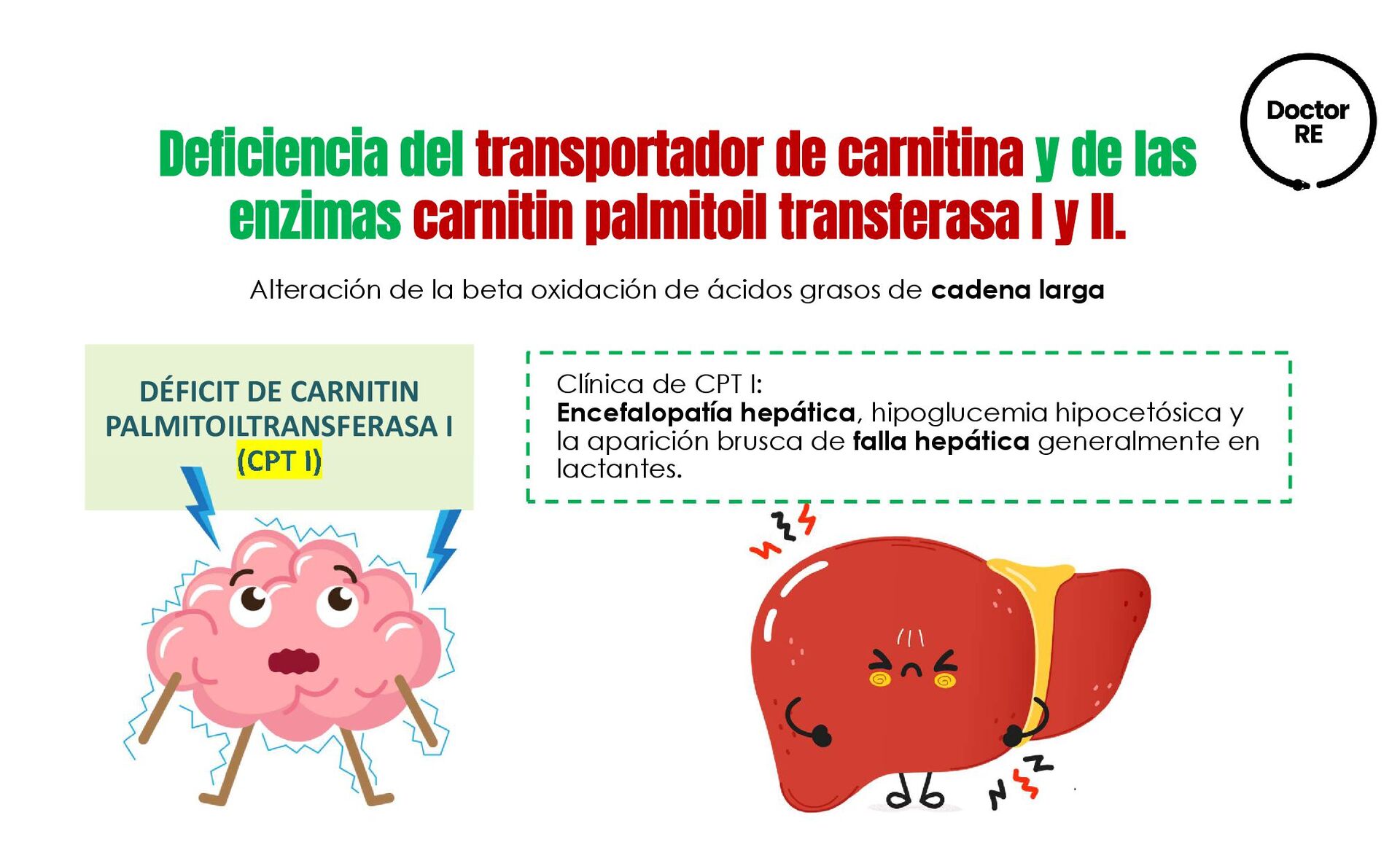
larga Clínica de CPT I: Encefalopatía hepática, hipoglucemia hipocetósica y la aparición brusca de falla hepática generalmente en lactantes. DÉFICIT DE CARNITIN PALMITOILTRANSFERASA I (CPT I) Deficiencia del transportador de carnitina y de las enzimas carnitin palmitoil transferasa I y II.
larga Existen 3 formas: •Letal neonatal: los niños desarrollan insuficiencia respiratoria, convulsiones, insuficiencia hepática, miocardiopatía, arritmia, hipoglucemia hipocetósica •Hepatocardiomuscular infantil grave: Inicia en el 1er año de vida, afecta el hígado, corazón y los músculos. •Miopática: menos grave más frecuente, se caracteriza por episodio de mialgia, debilidad muscular y rabdomiólisis, pueden ser desencadenados por el ejercicio, el estrés, la exposición a temperaturas extremas, infecciones o el ayuno DÉFICIT DE CARNITIN PALMITOILTRANSFERASA II (CPT II) Deficiencia del transportador de carnitina y de las enzimas carnitin palmitoil transferasa I y II.
en tandem • Diagnóstico definitivo: detección de la reducción de actividad de la enzima TRATAMIENTO • De la carnitina-acilcarnitina translocasa y de carnitina palmitoil transferasa tipo II, se indica 150 a 200 mg/kg/día de L carnitina • Deficiencia de CPT II es dietético con administración de carbohidratos (70%) y grasa (<20%) RECOMENDACIONES • Se recomienda evitar ayuno por más de 4 horas en niños de 4 meses, en niños de 5 y 12 meses se puede agregar una hora por mes hasta máximo 8 horas de ayuno • Comidas frecuentes • Incrementar la ingestión de hidratos de carbono durante los episodios en los que el requerimiento energético sea mayor • Niños entre 4 y 8 años de edad deben consumir 10g de ácido linoleico y 0.9g de ácido α- linolénico por día
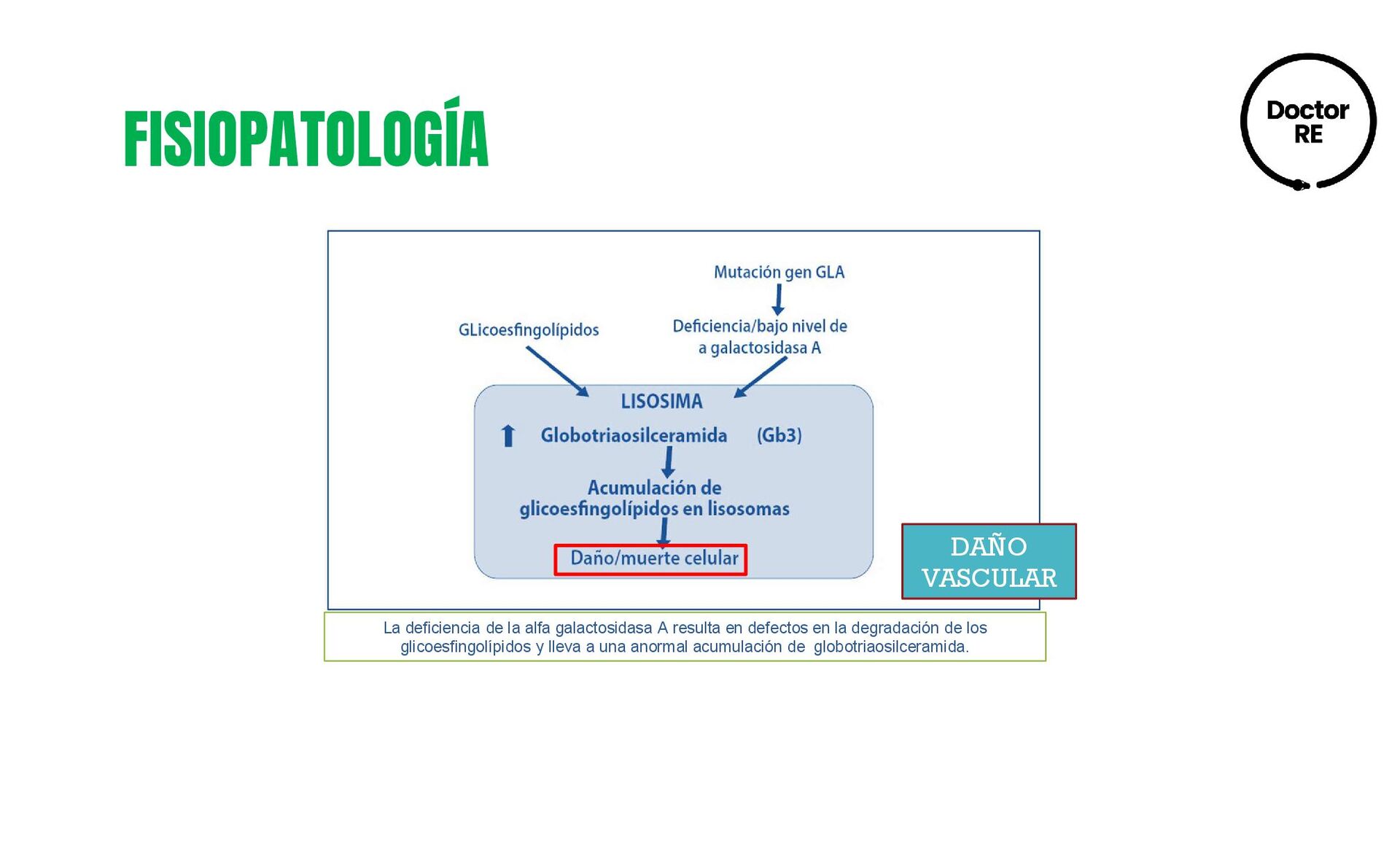
la AGLA, situado en el cromosoma X (Xq22.1), que originan un déficit de Alfa A-galactasidasa lo que condiciona la acumulación de glucoesfingolípidos en los lisosomas. • La causa de muerte es en primer lugar insuficiencia renal; en segundo insuficiencia cardíaca, y accidentes cerebrovasculares. • La incidencia a nivel global es cercana a 1:3.100 en varones, se desconoce la cifra de ambos géneros. • Prevalencia de la variante renal es de 0.2-1.2% • Prevalencia de la variante cardiaca es de 1-6.3% • Prevalencia de la variante cerebrovascular es 4.9% en varones y 2.4% en mujeres DEFINICIÓN
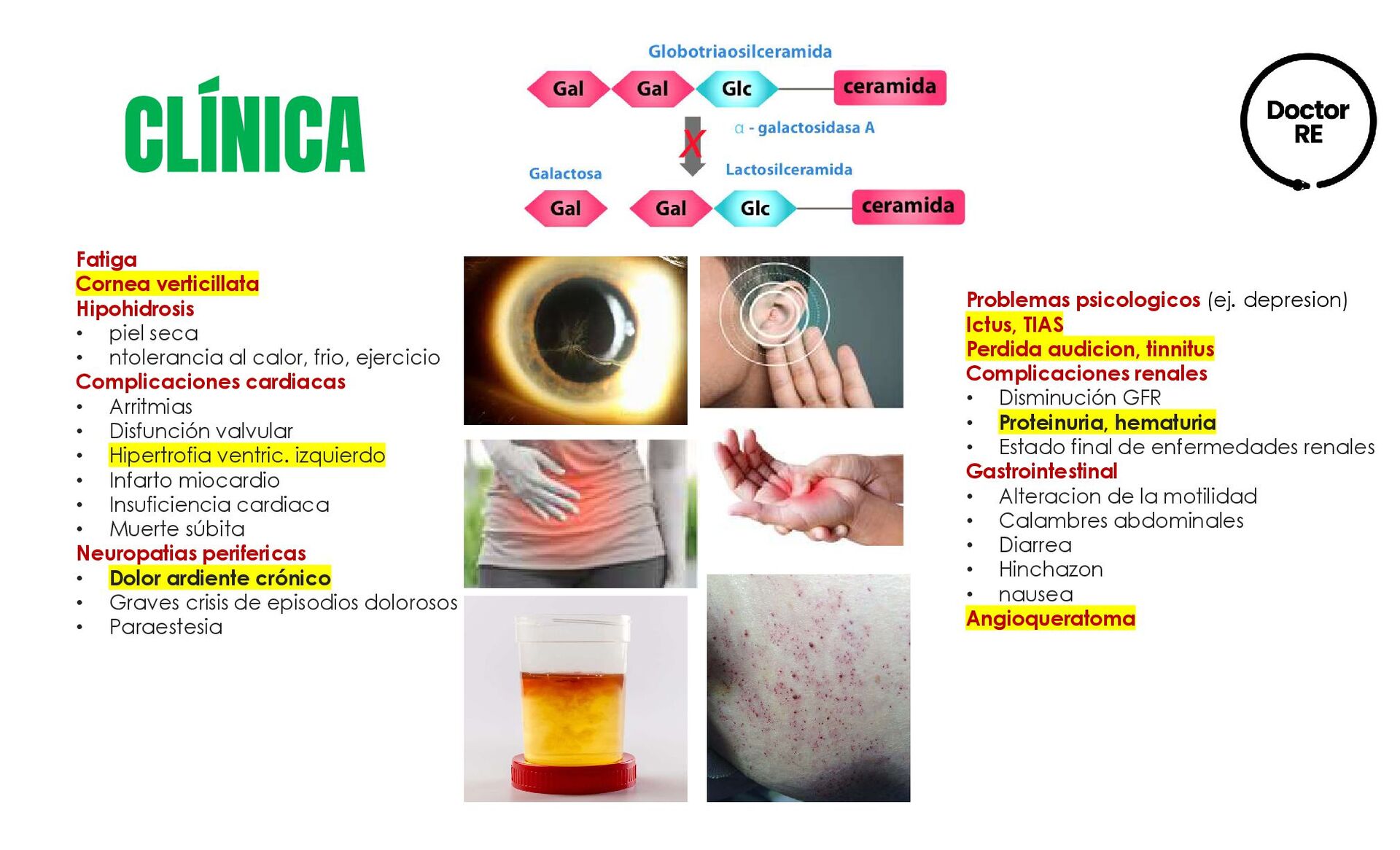
neuropático, dolores crónicos en las articulaciones, anomalías oftalmológicas, hipoacusia, hipersensibilidad al calor y el frío, trastornos gastrointestinales, letargo y fatiga, angioqueratomas, primeras anomalías renales y cardiacas. Edad adulta temprana (17-30 años). • Otros angioqueratomas, linfedemas en MP, riñones: proteinuria e IR avanzada • Corazón: miocardiopatía hipertrófica e hipertrofia de ventrículo izquierdo, angina de pecho, arritmias • SNC: ataque isquémico transitorio, infarto cerebral, depresión. Edad adulta avanzada (>30 años). • Insuficiencia cardiaca, trastornos del ritmo cardiaco • Recidiva de AIT e infarto cerebral • Insuficiencia renal, necesidad de diálisis
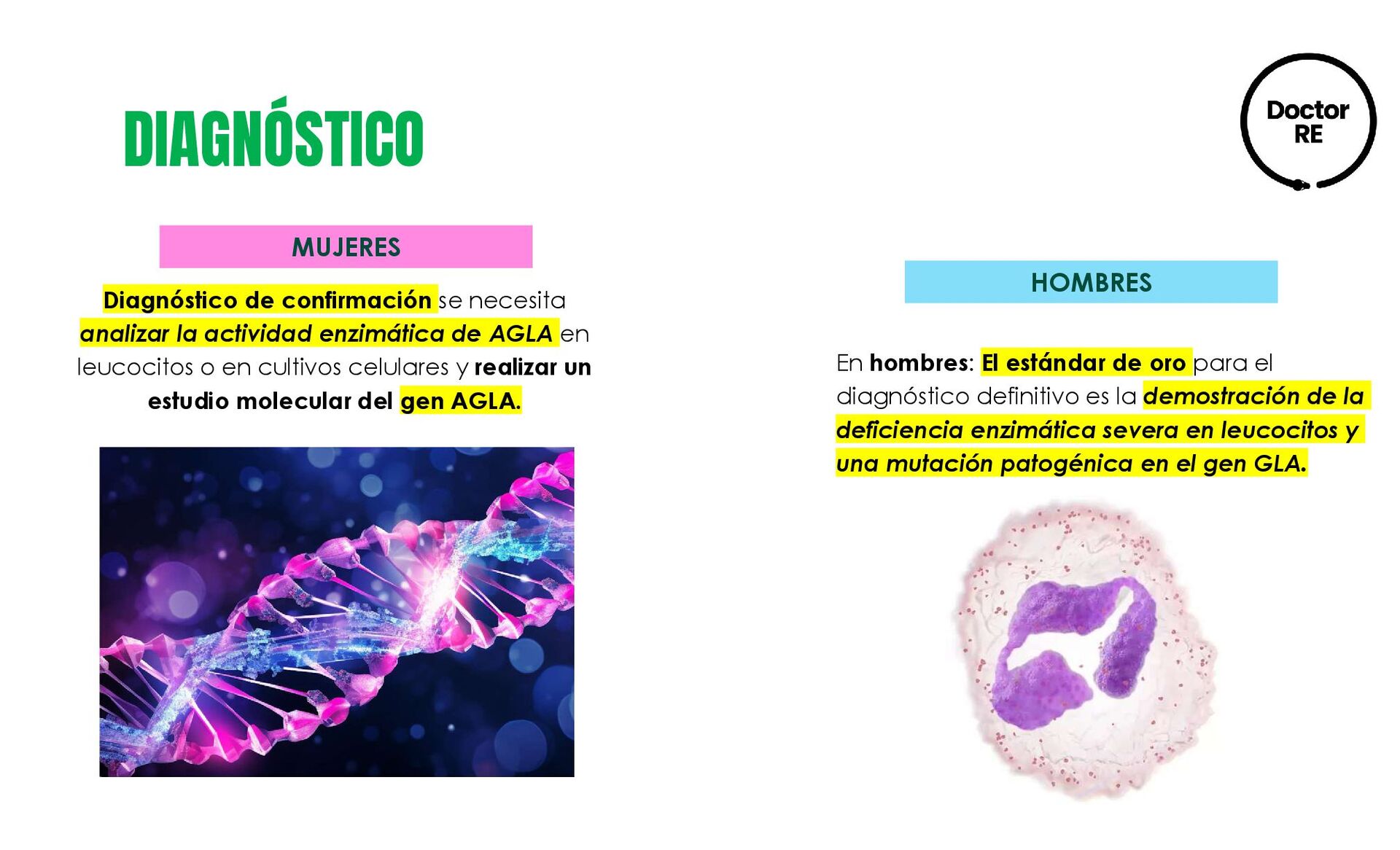
de AGLA en leucocitos o en cultivos celulares y realizar un estudio molecular del gen AGLA. En hombres: El estándar de oro para el diagnóstico definitivo es la demostración de la deficiencia enzimática severa en leucocitos y una mutación patogénica en el gen GLA. MUJERES HOMBRES
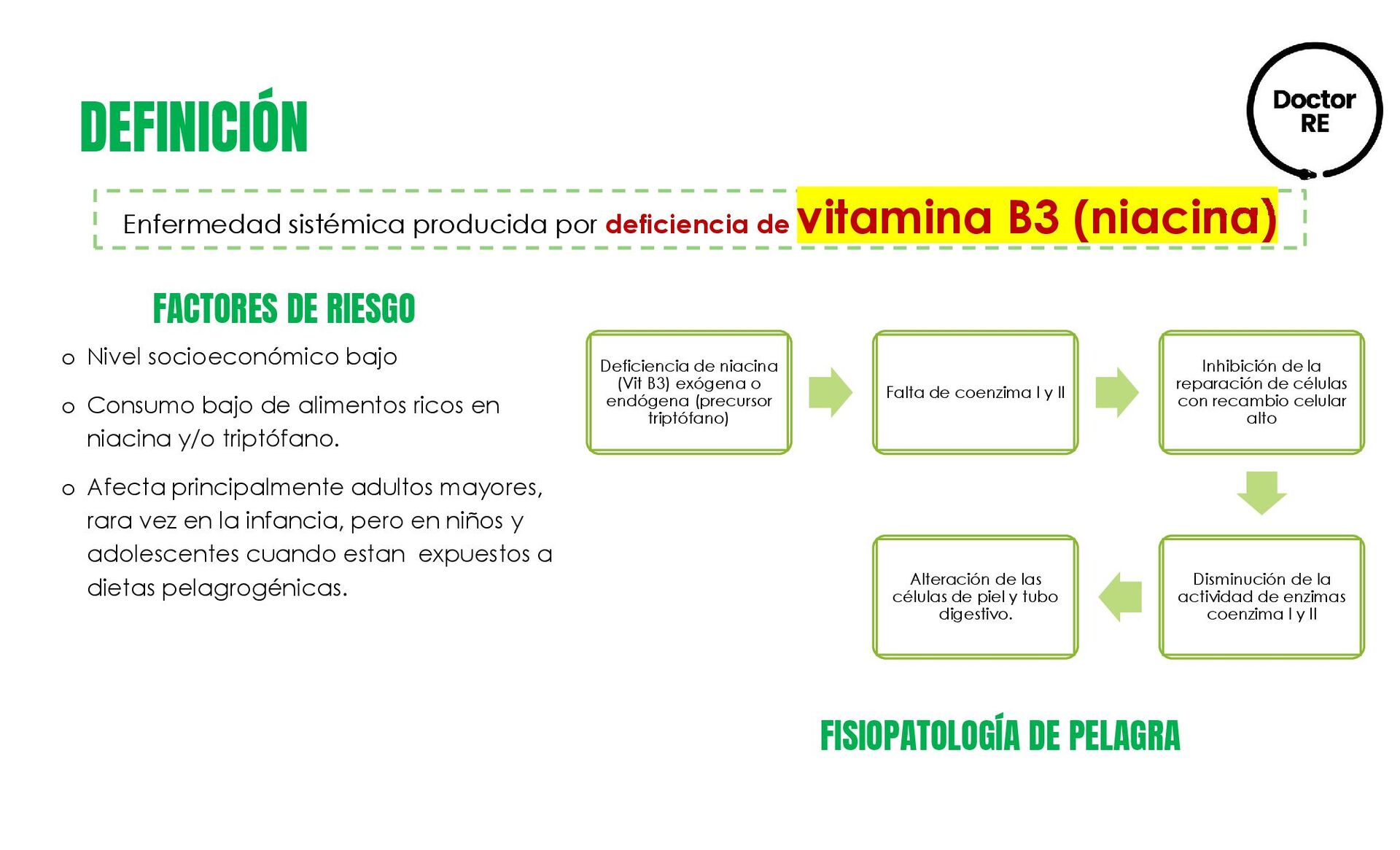
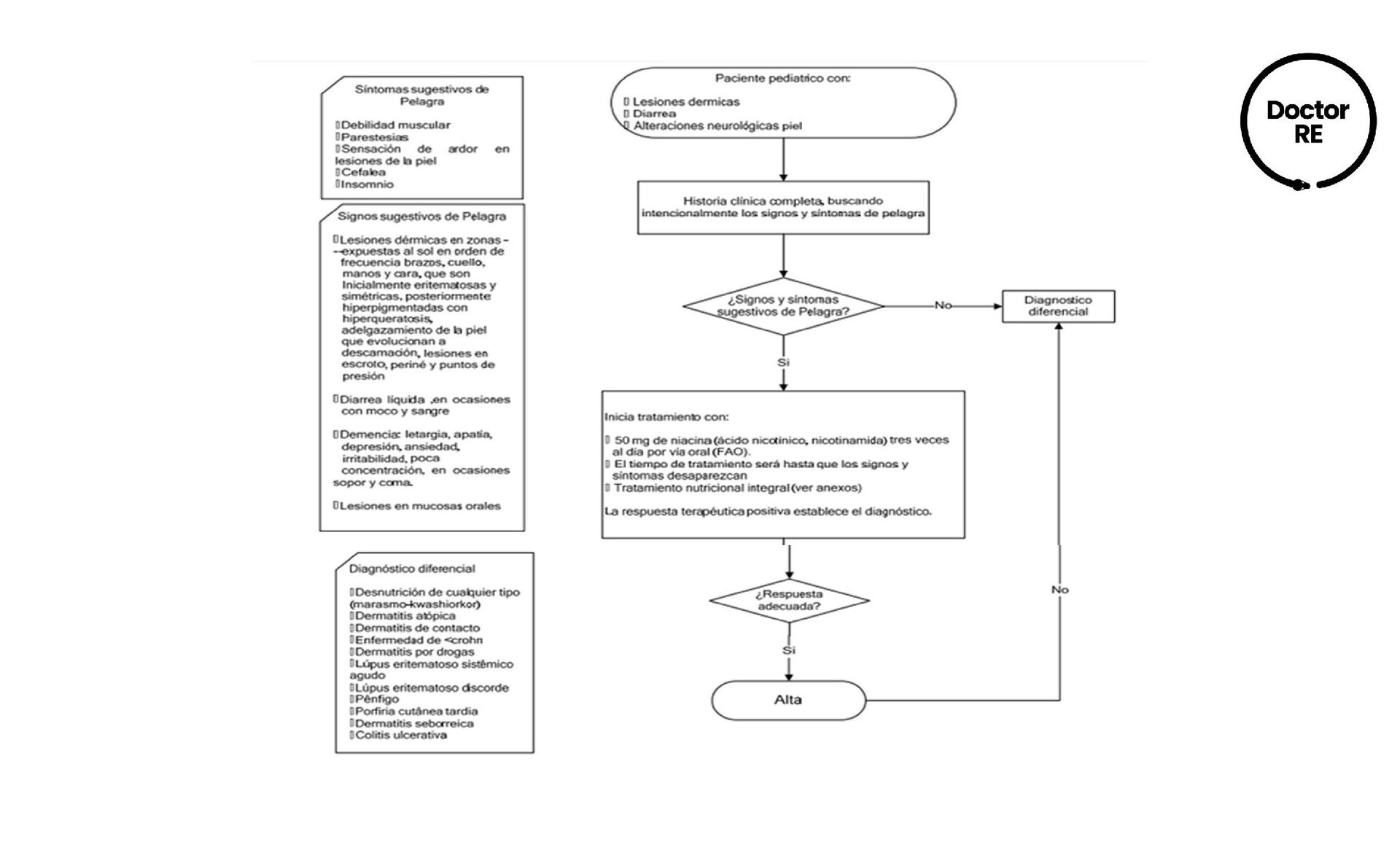
Nivel socioeconómico bajo o Consumo bajo de alimentos ricos en niacina y/o triptófano. o Afecta principalmente adultos mayores, rara vez en la infancia, pero en niños y adolescentes cuando estan expuestos a dietas pelagrogénicas. Deficiencia de niacina (Vit B3) exógena o endógena (precursor triptófano) Falta de coenzima I y II Inhibición de la reparación de células con recambio celular alto Disminución de la actividad de enzimas coenzima I y II Alteración de las células de piel y tubo digestivo. FISIOPATOLOGÍA DE PELAGRA DEFINICIÓN FACTORES DE RIESGO
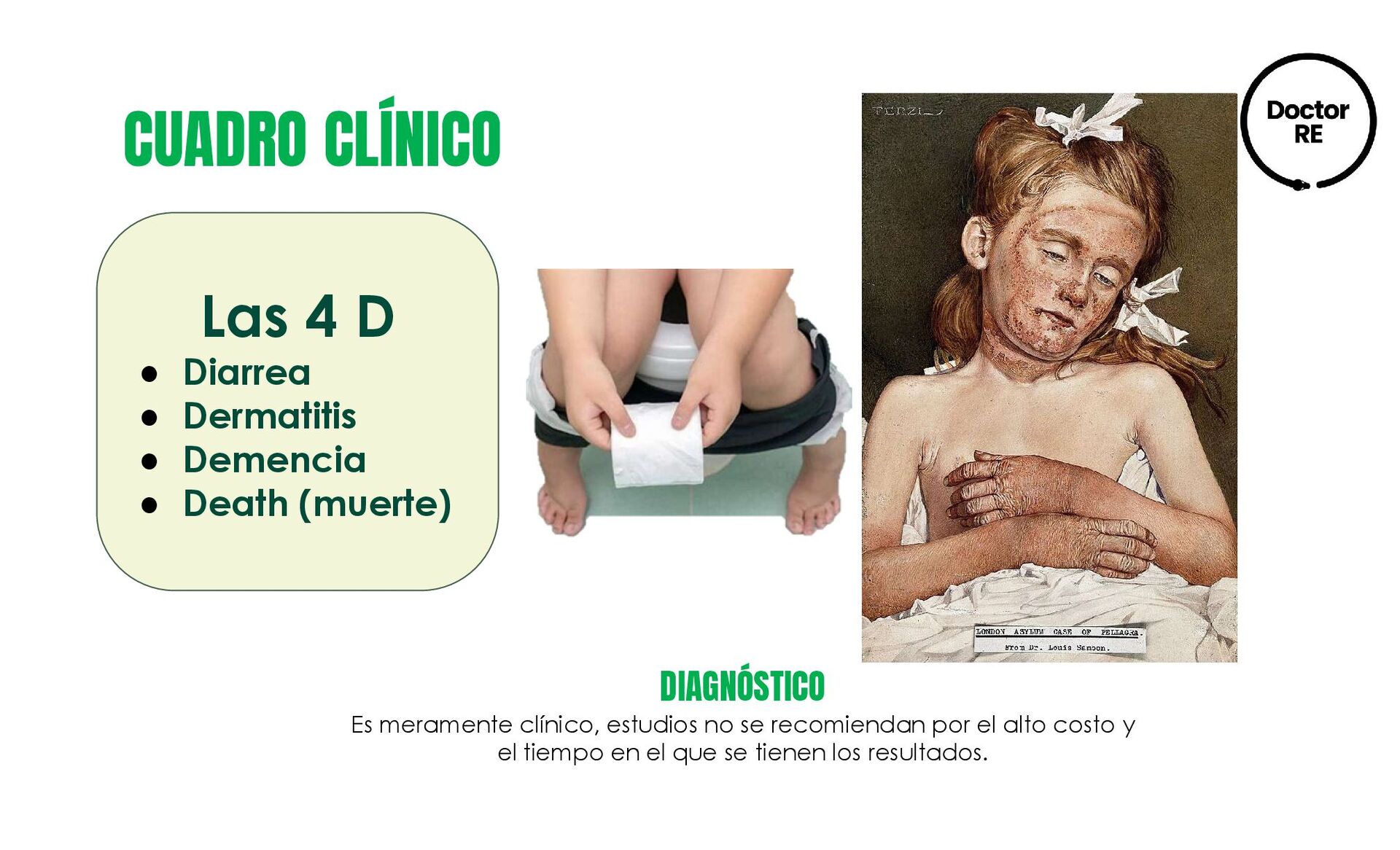
Demencia • Death (muerte) DIAGNÓSTICO Es meramente clínico, estudios no se recomiendan por el alto costo y el tiempo en el que se tienen los resultados.
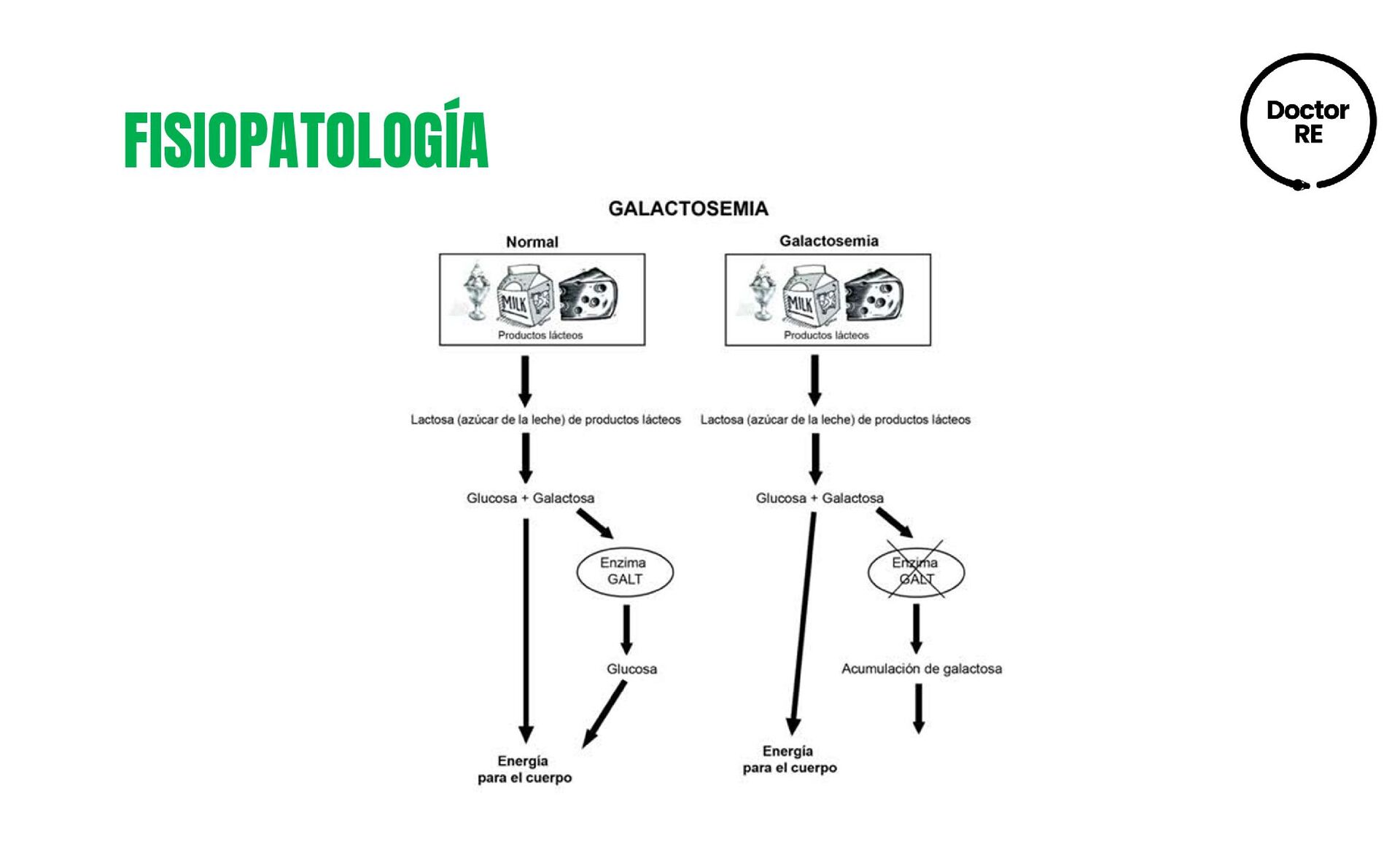
carbono ocasionada por una deficiencia enzimática, lo que resulta en la acumulación de los metabolitos galactitol y galactosa-1-fosfato DEFINICIÓN ETIOLOGÍA • Mutaciones en el gen GALT. • Las mutaciones más frecuentes México en un 71% de los casos son la Q188R, IVS2-2A>G y N314D • Más severas por presentar ausencia de actividad enzimática. Frecuencia 1:14,000 a 1:80,000 a nivel mundial. 1:59 -158 en México
1. Galactosemia causada por la deficiencia de galactosa-1-fosfato uridil transferasa (GALT) y se clasifica en: °Galactosemia clásica °Galactosemia clínica °Galactosemia bioquímica Tipo 2. Deficiencia de galactosa cinasa (GALK/GALK). Tipo 3. Deficiencia de galactosa-4-fosfato epimerasa. Galactosemia secundaria Generada por patologías como: hepatitis congénita, malformaciones hepáticas arteriovenosas, alteraciones metabólicas como tirosinemia tipo I, citrulinemia tipo 2 y síndrome de Fanconi- Bickel.
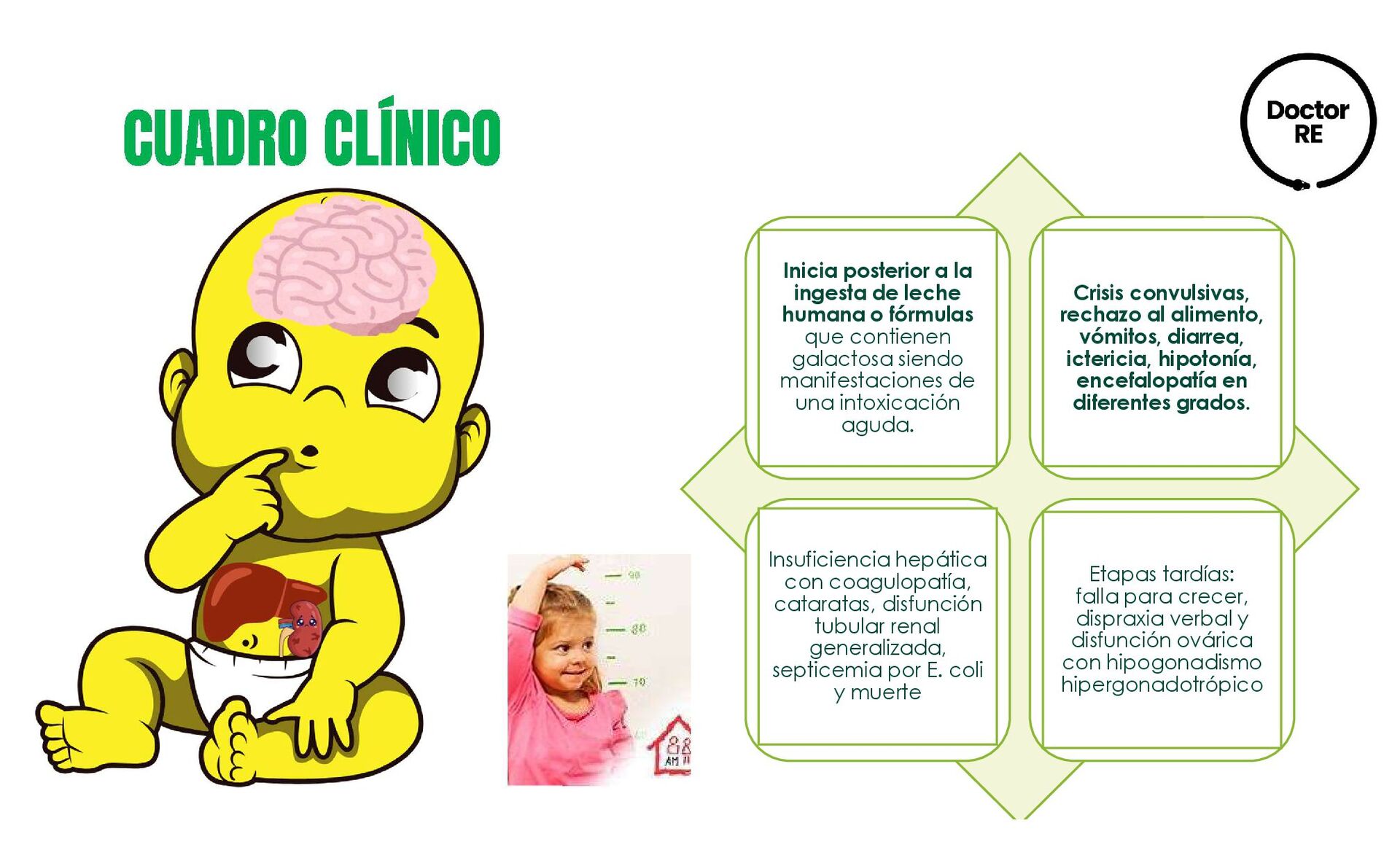
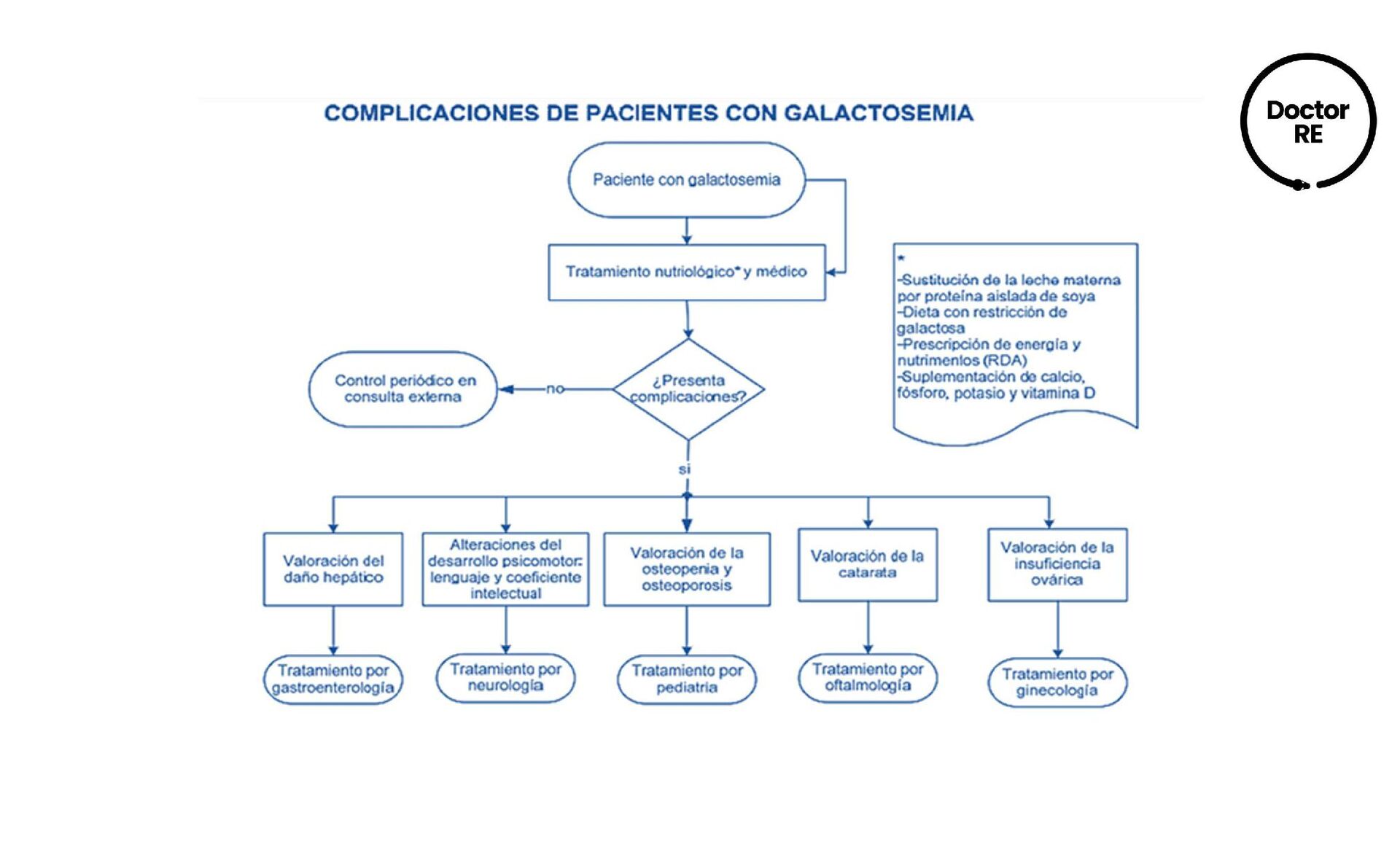
o fórmulas que contienen galactosa siendo manifestaciones de una intoxicación aguda. Crisis convulsivas, rechazo al alimento, vómitos, diarrea, ictericia, hipotonía, encefalopatía en diferentes grados. Insuficiencia hepática con coagulopatía, cataratas, disfunción tubular renal generalizada, septicemia por E. coli y muerte Etapas tardías: falla para crecer, dispraxia verbal y disfunción ovárica con hipogonadismo hipergonadotrópico
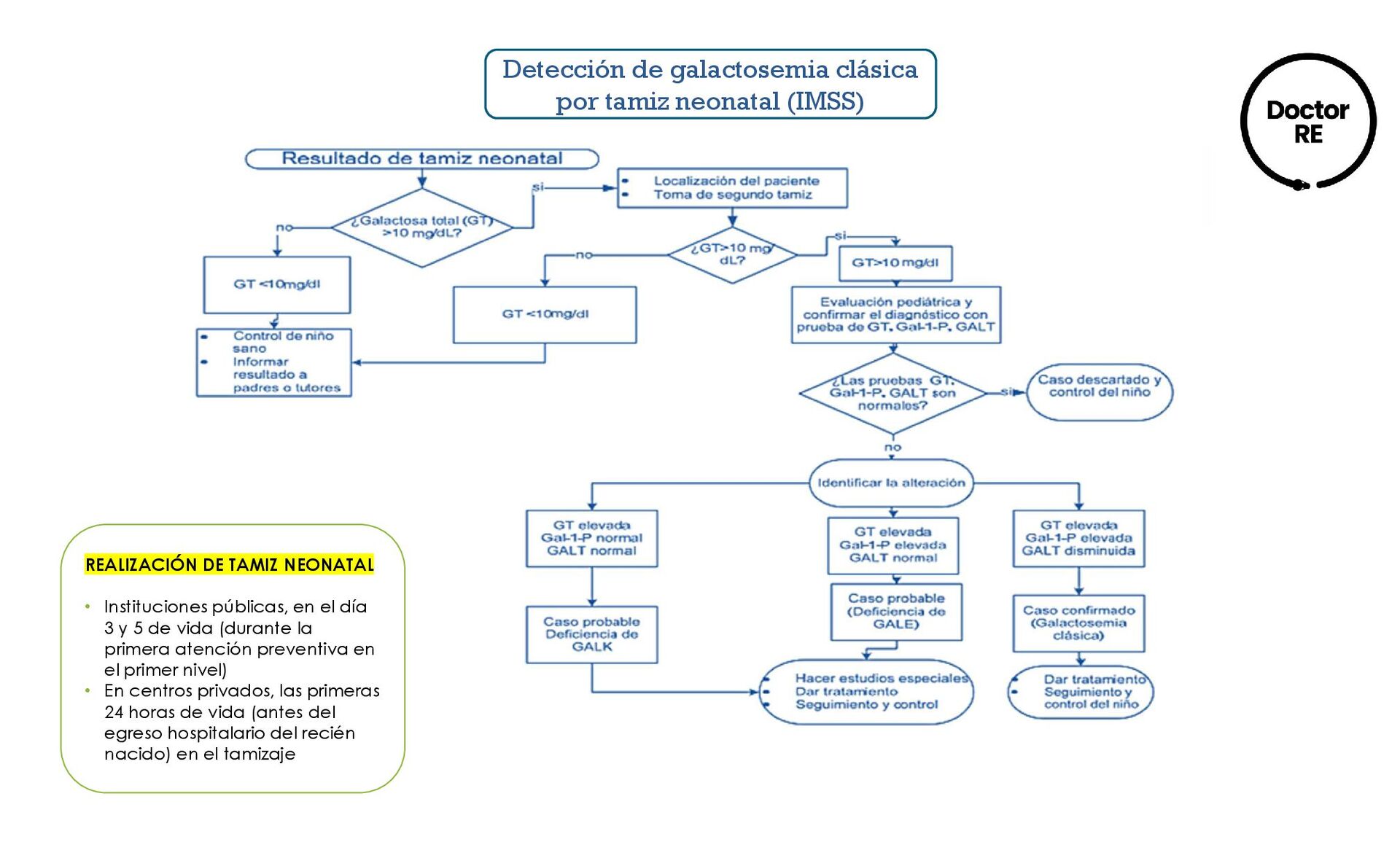
(GT) y G-1-P • Se establece cuando los resultados de galactosa total es mayor o igual a 10 mg/dl en el tamiz neonatal. DIAGNÓSTICO DEFINITIVO • Medición de galactosa uridiltransferasa (GALT) y galactosa-1-fosfato en plasma por reacción enzimática, ensayo espectrofotométrico y/o análisis cuantitativo radioenzimático. • Galactosa-uridiltransferasa (GALT) menor o igual a 9.5 umol/h/gr Hb (Duarte). Menor a 2 umol/h/gr Hb (clásica). • Galactosa-1-fosfato (GALT-1-P) mayor o igual a 1 mg/dl
TAMIZ NEONATAL • Instituciones públicas, en el día 3 y 5 de vida (durante la primera atención preventiva en el primer nivel) • En centros privados, las primeras 24 horas de vida (antes del egreso hospitalario del recién nacido) en el tamizaje
y mantenerse de un modo estricto durante toda la vida mediante: ▪ Eliminación de la galactosa de la dieta, ante la menor sospecha clínica o resultado positivo de las pruebas confirmatorias para prevenir las secuelas irreversibles o muerte en la etapa neonatal. ▪ Fórmula de proteína de soya. ▪ Micronutrimentos, vitamina D y calcio
Oxidación De Ácidos Grasos (errores innatos del metabolismo) Población pediátrica Nivel de Atención: 1º, 2º y 3º, México: Secretaría de Salud. ▪ Diagnóstico, tratamiento y seguimiento multidisciplinario en enfermedad de Fabry en el segundo y tercer nivel de atención. Ciudad de México, Secretaría de Salud. ▪ Diagnóstico y Tratamiento de la Pelagra en Edad Pediátrica. Guía de Evidencias y Recomendaciones: Guía de Práctica Clínica. México, CENETEC. ▪ Galactosemia. Tamizaje, diagnóstico, tratamiento médico de las complicaciones e intervención nutricional. Guía de Evidencias y Recomendaciones: Guía de Práctica Clínica. México, CENETEC.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}