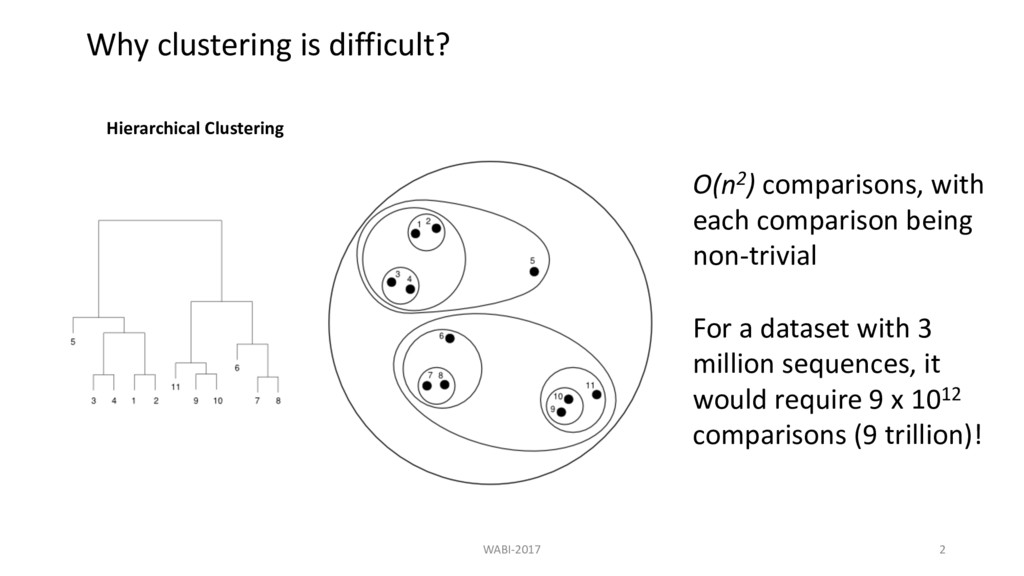
many DNA sequences within 5 mismatches in first 500 bp and one mismatch in last position? 3 " 3# " 500 5 ≈ 95 " 10*+ Filtering methods based on kmers won’t help!
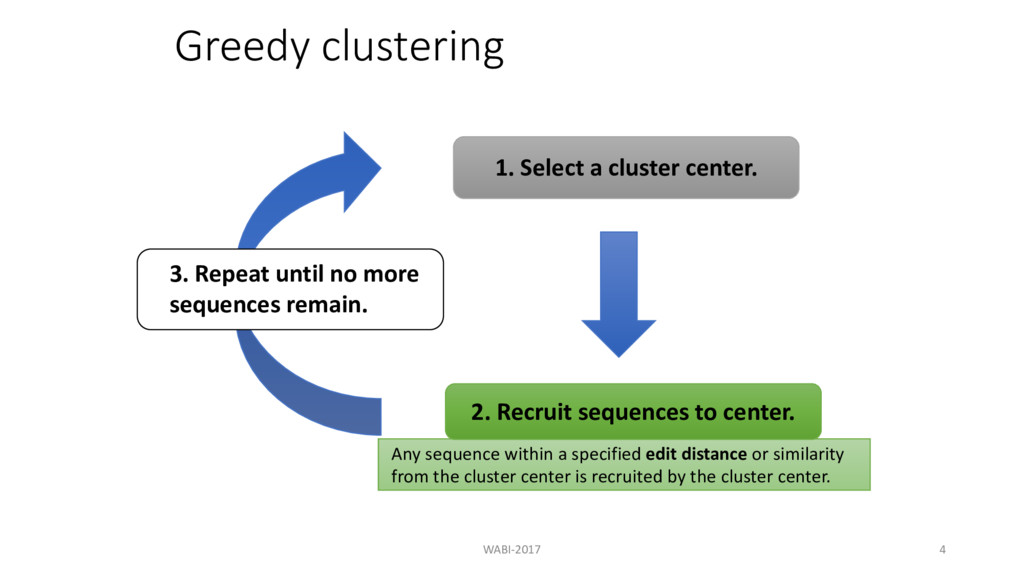
the cluster center is recruited by the cluster center. 1. Select a cluster center. 2. Recruit sequences to center. 3. Repeat until no more sequences remain. Greedy clustering WABI-2017 4
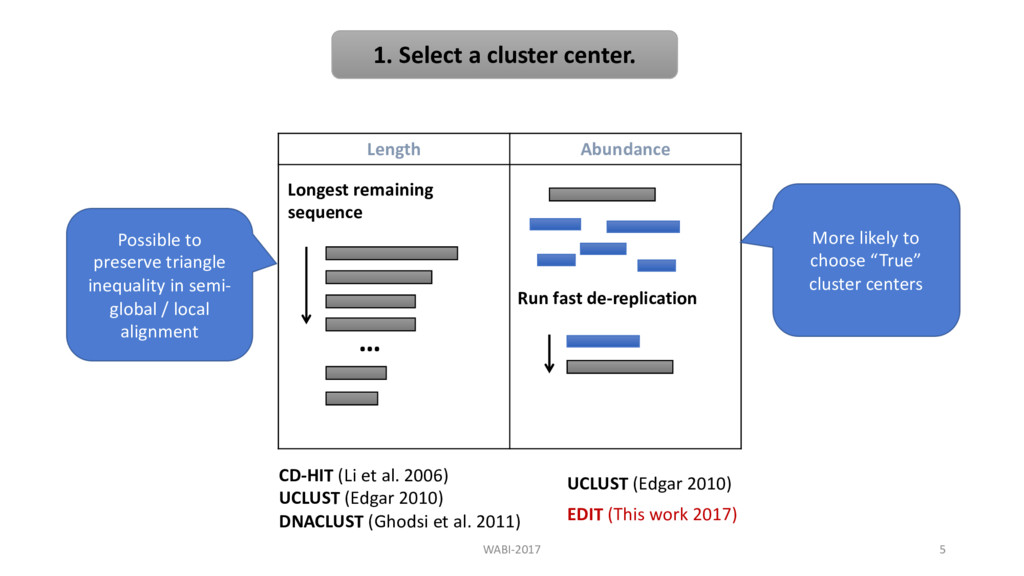
de-replication CD-HIT (Li et al. 2006) UCLUST (Edgar 2010) DNACLUST (Ghodsi et al. 2011) UCLUST (Edgar 2010) Longest remaining sequence EDIT (This work 2017) WABI-2017 5 Possible to preserve triangle inequality in semi- global / local alignment More likely to choose “True” cluster centers
Solution Dynamic programming Takes O(m2) time where m is the length of the sequence. The Strong Exponential Time Hypothesis (SETH) implies it cannot be done faster (Backrus and Indyk, 2015) WABI-2017 7
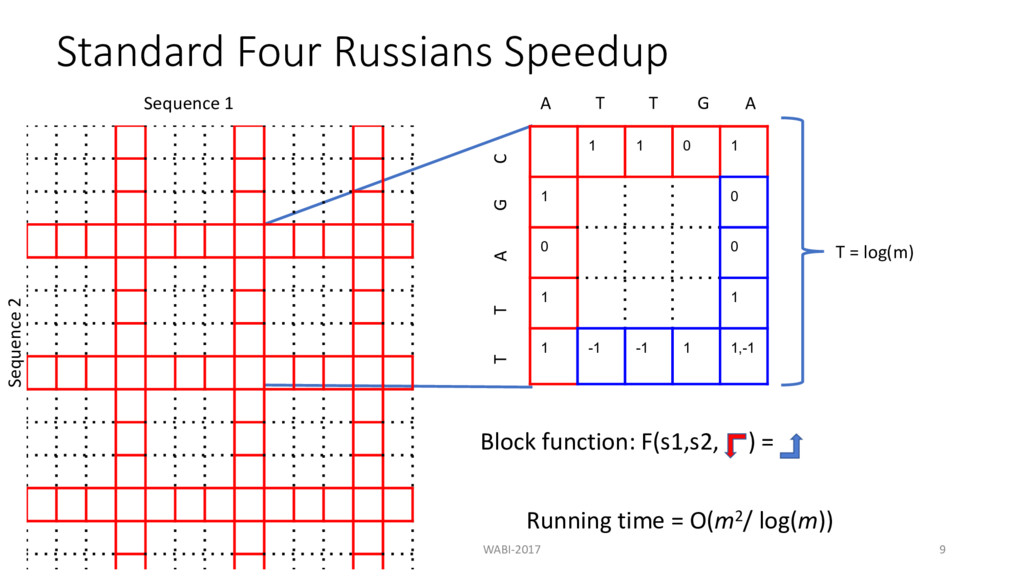
, - − 1, / + 1 , -, / − 1 + 1 , - − 1, / − 1 + (8 - == 9 / ? 0 ∶ 1) Theorem: Under standard formulation, any two adjacent cells in edit distance matrix differ by atmost 1. Hence the possible values the difference between adjacent cells can take are 0, 1 and -1. (Gusfield, 1997)
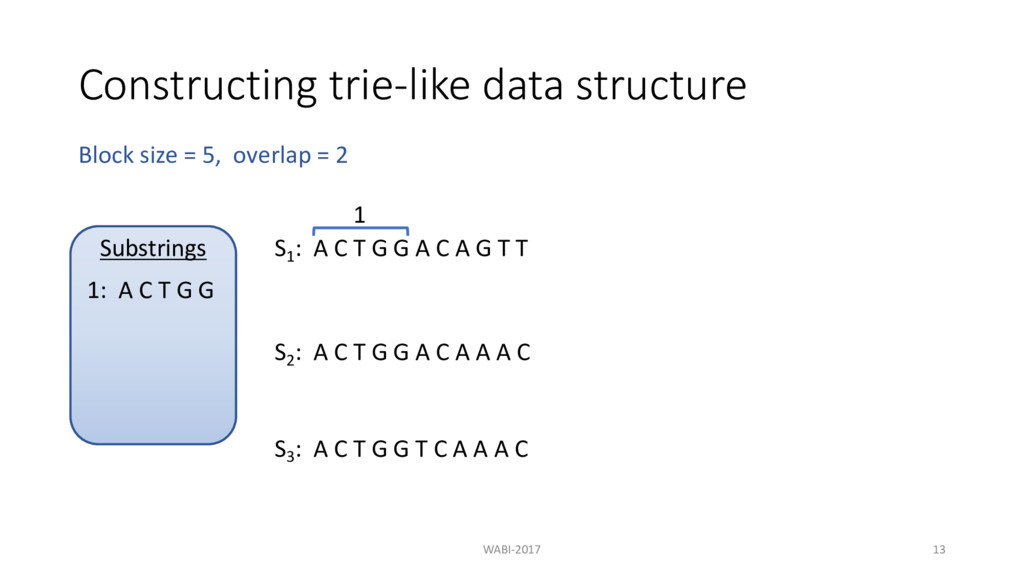
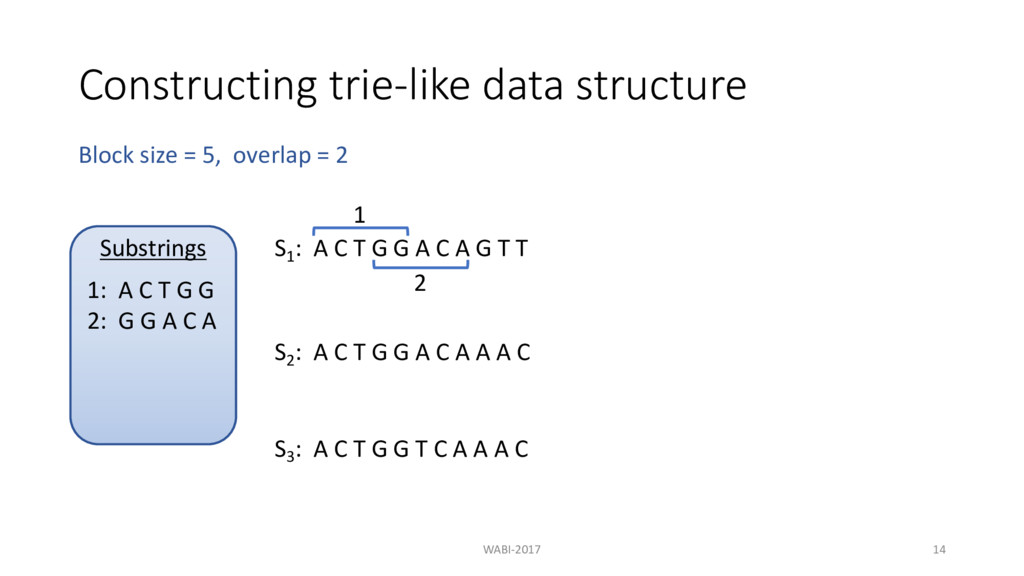
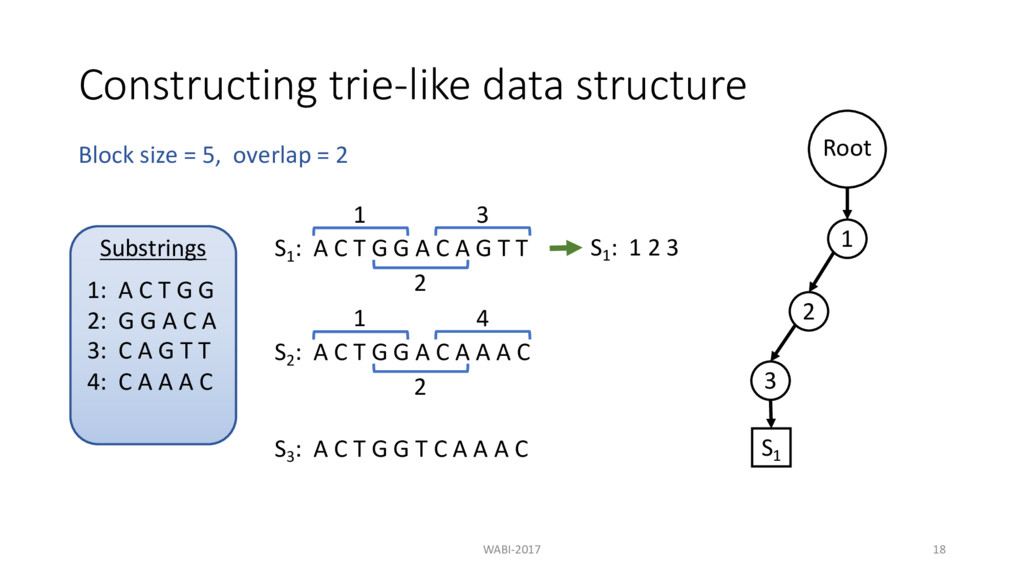
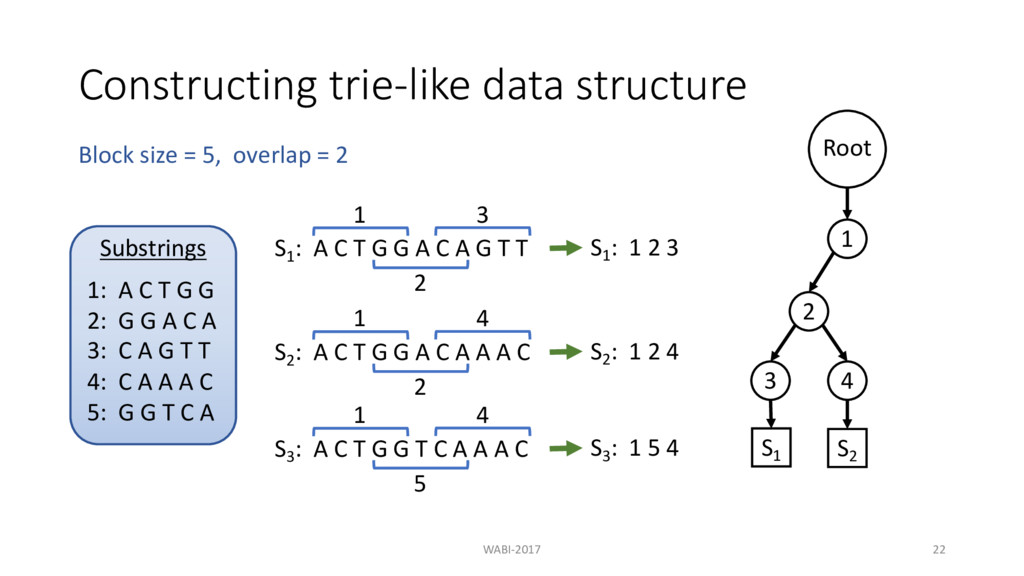
T G G A C A G T T 1 2 3 S2 : A C T G G A C A A A C S3 : A C T G G T C A A A C Block size = 5, overlap = 2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T
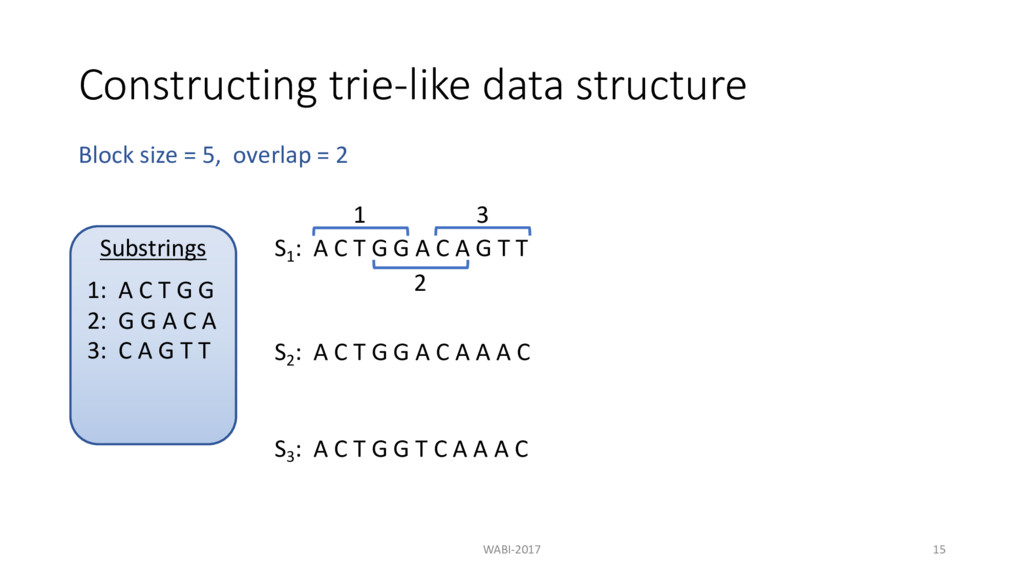
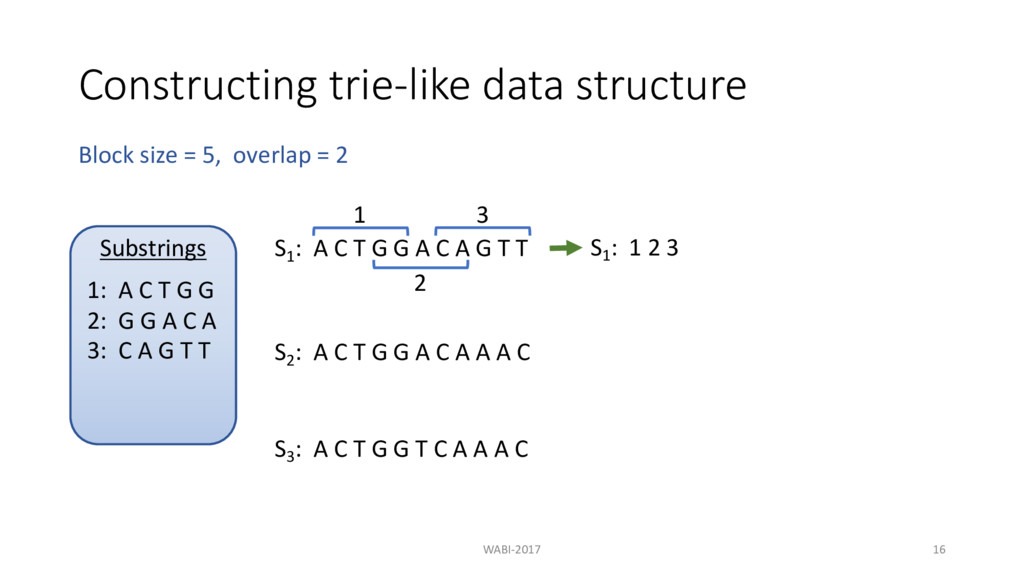
T G G A C A G T T 1 S1 : 1 2 3 2 3 S2 : A C T G G A C A A A C S3 : A C T G G T C A A A C Block size = 5, overlap = 2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T
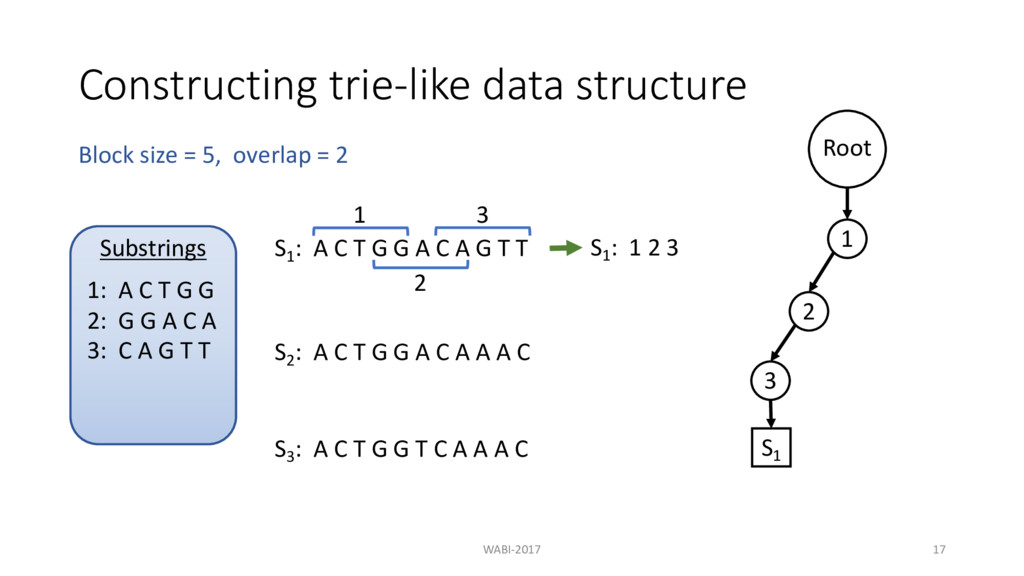
T G G A C A G T T 1 S1 : 1 2 3 2 3 S2 : A C T G G A C A A A C S3 : A C T G G T C A A A C Block size = 5, overlap = 2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T Root 1 2 3 S1
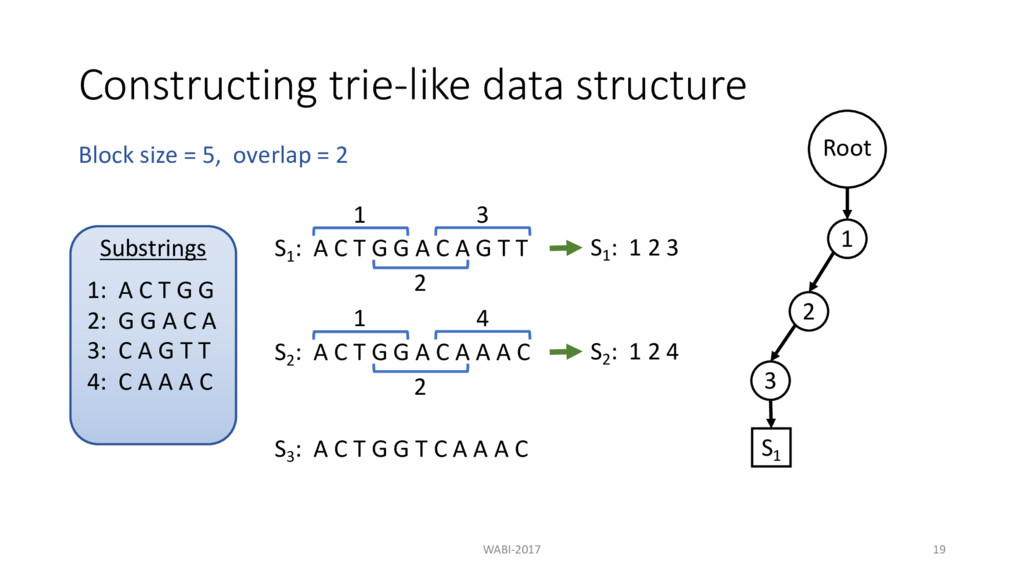
T G G A C A G T T 1 S1 : 1 2 3 2 3 S2 : A C T G G A C A A A C 1 2 4 S3 : A C T G G T C A A A C Block size = 5, overlap = 2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C Root 1 2 3 S1
T G G A C A G T T 1 S1 : 1 2 3 2 3 S2 : A C T G G A C A A A C 1 2 4 S3 : A C T G G T C A A A C Block size = 5, overlap = 2 S2 : 1 2 4 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C Root 1 2 3 S1
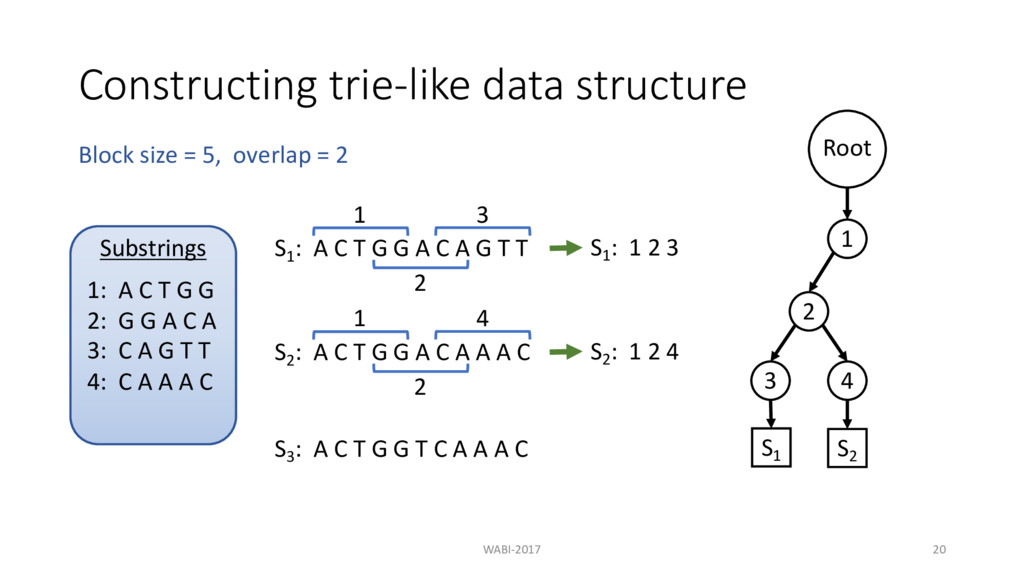
T G G A C A G T T 1 S1 : 1 2 3 2 3 S2 : A C T G G A C A A A C 1 2 4 S3 : A C T G G T C A A A C Block size = 5, overlap = 2 S2 : 1 2 4 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C Root 1 2 3 4 S1 S2
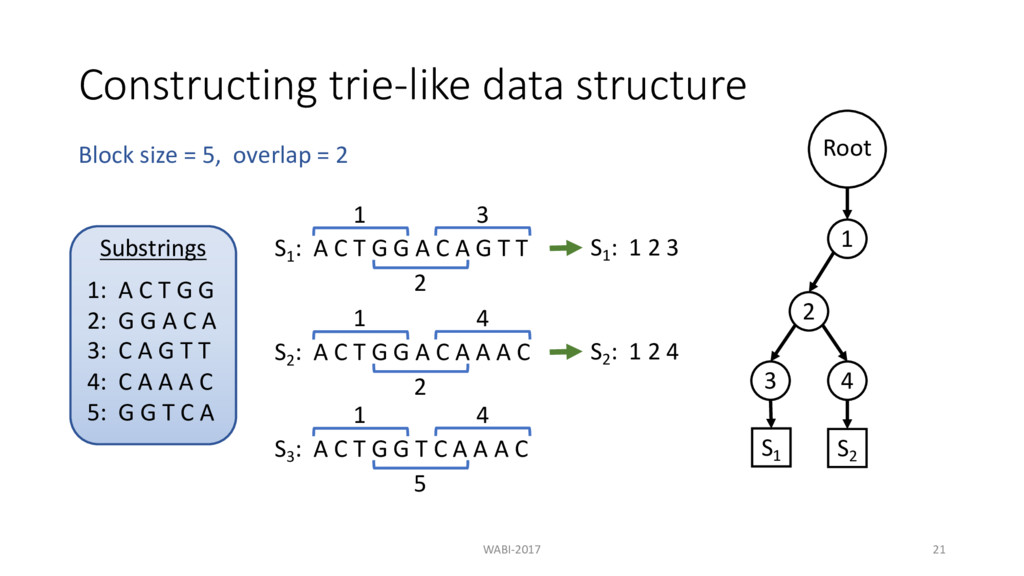
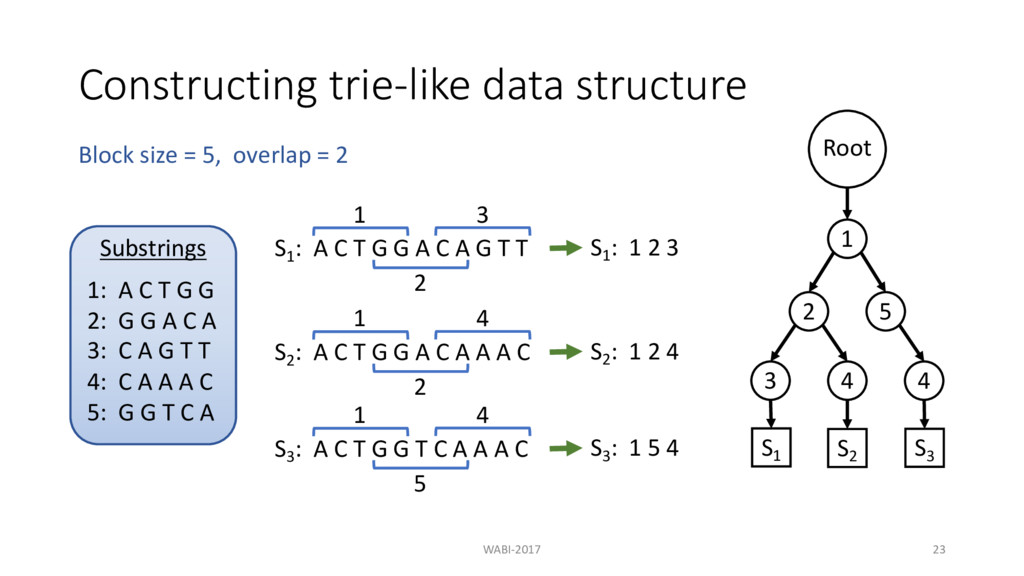
T G G A C A G T T 1 S1 : 1 2 3 2 3 S2 : A C T G G A C A A A C 1 2 4 S3 : A C T G G T C A A A C 1 5 4 Block size = 5, overlap = 2 S2 : 1 2 4 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C 5: G G T C A Root 1 2 3 4 S1 S2
T G G A C A G T T 1 S1 : 1 2 3 2 3 S2 : A C T G G A C A A A C 1 2 4 S3 : A C T G G T C A A A C 1 5 4 Block size = 5, overlap = 2 S2 : 1 2 4 S3 : 1 5 4 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C 5: G G T C A Root 1 2 3 4 S1 S2
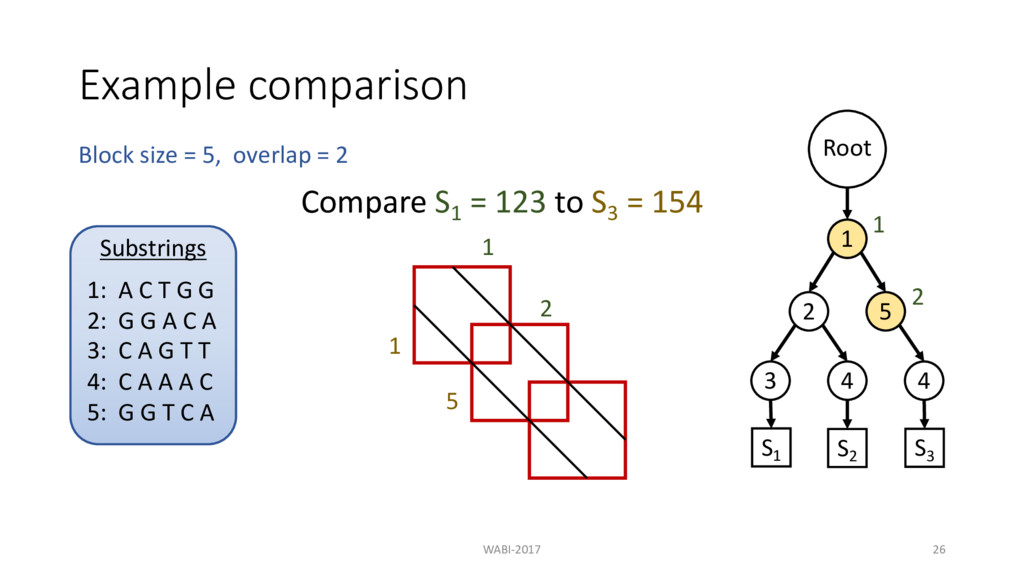
T G G A C A G T T 1 S1 : 1 2 3 2 3 S2 : A C T G G A C A A A C 1 2 4 S3 : A C T G G T C A A A C 1 5 4 Block size = 5, overlap = 2 S2 : 1 2 4 S3 : 1 5 4 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C 5: G G T C A Root 1 2 3 4 5 4 S1 S2 S3
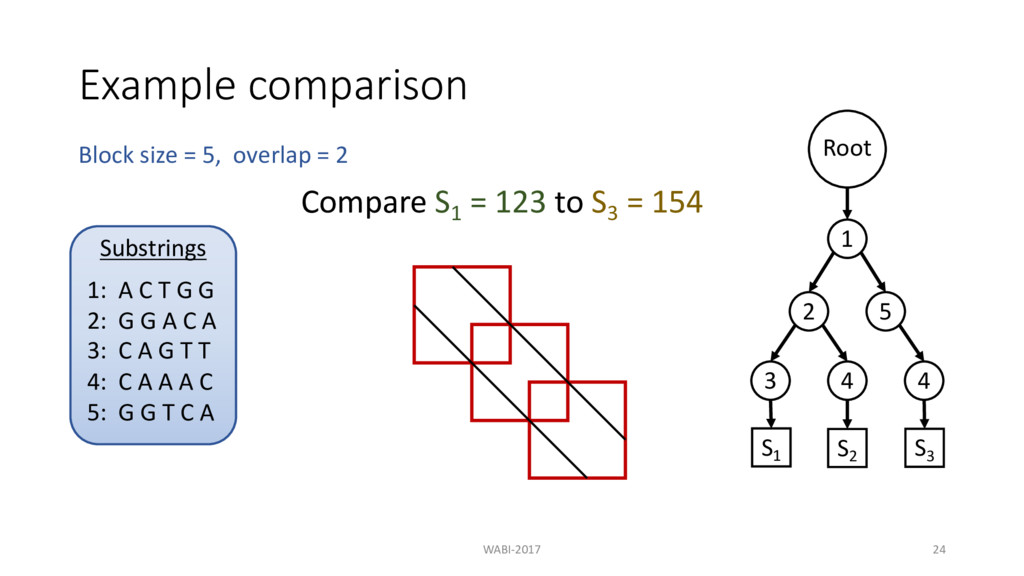
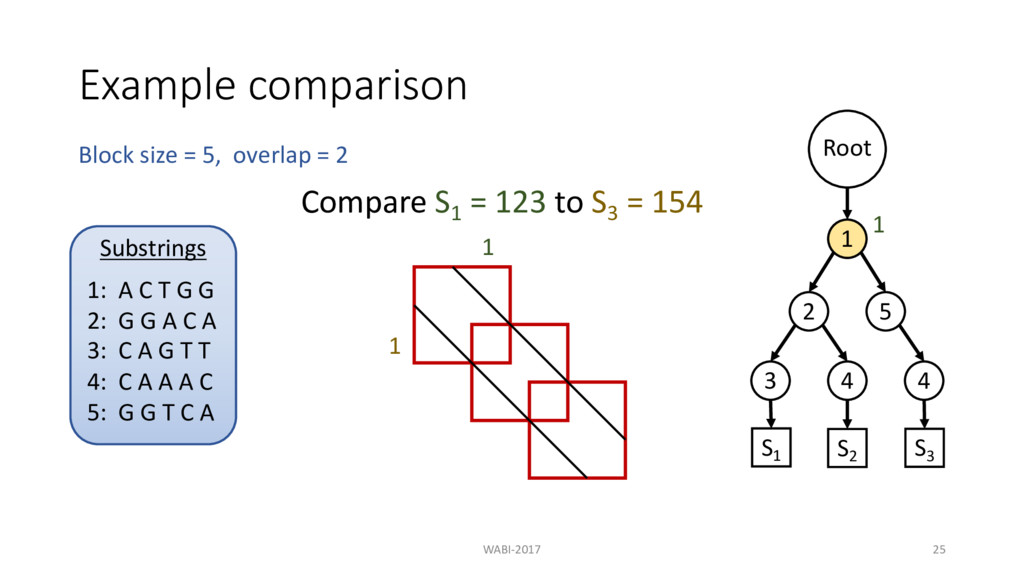
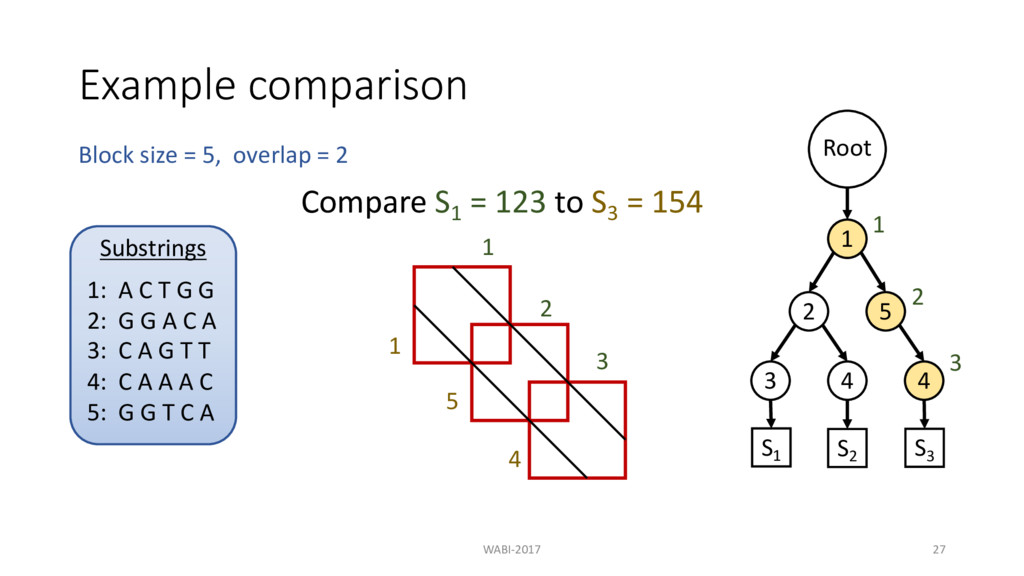
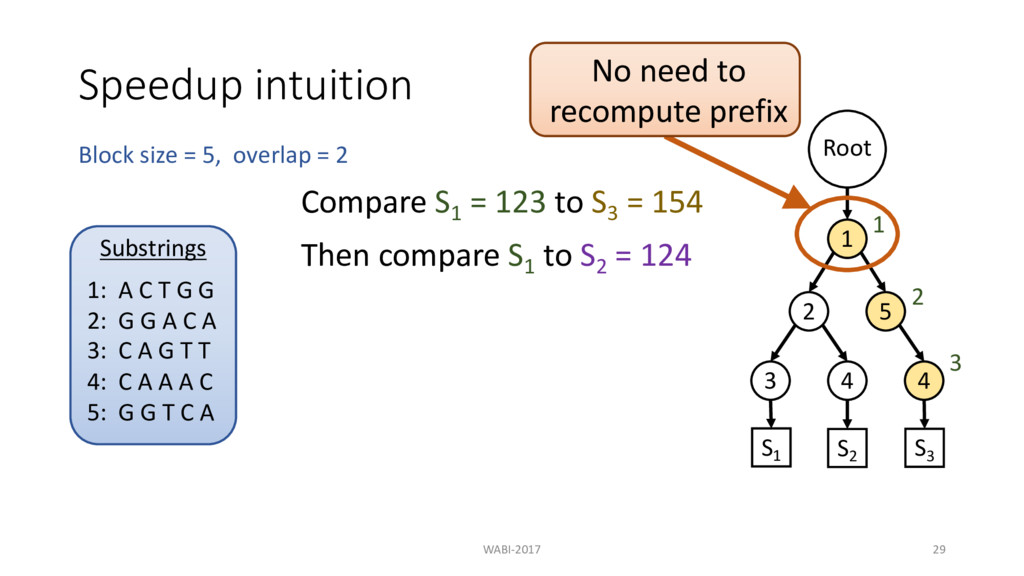
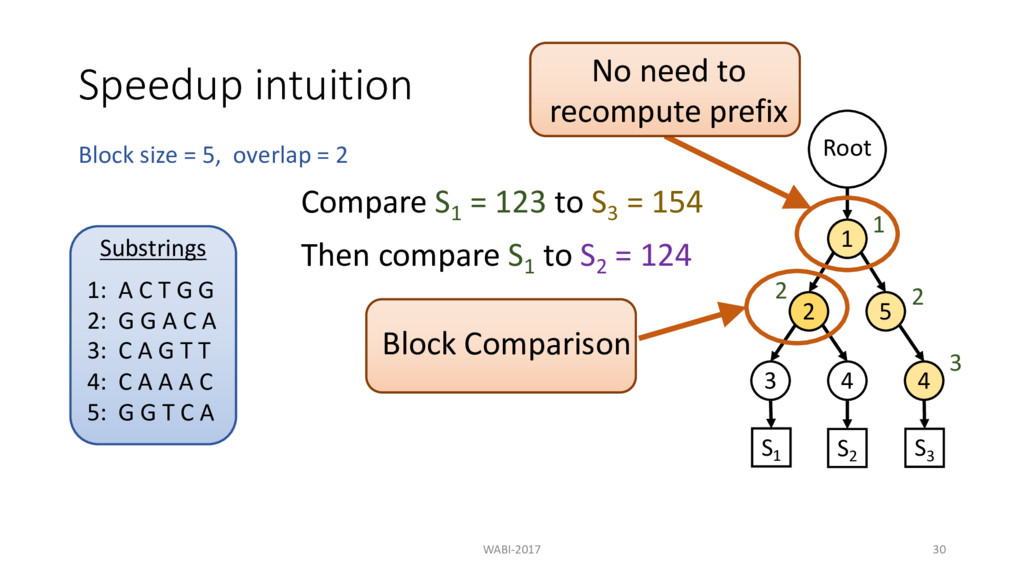
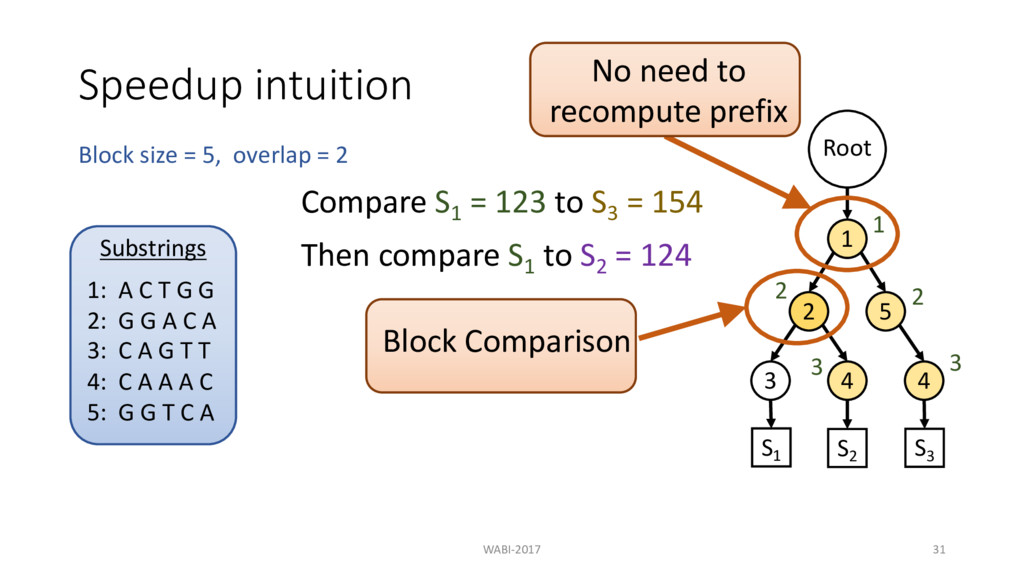
2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C 5: G G T C A Root 1 2 3 4 5 4 S1 S2 S3 Compare S1 = 123 to S3 = 154 Then compare S1 to S2 = 124 1 2 3
2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C 5: G G T C A Root 1 2 3 4 5 4 S1 S2 S3 Compare S1 = 123 to S3 = 154 Then compare S1 to S2 = 124 1 2 3 No need to recompute prefix
2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C 5: G G T C A Root 1 2 3 4 5 4 S1 S2 S3 Compare S1 = 123 to S3 = 154 Then compare S1 to S2 = 124 1 2 3 2 No need to recompute prefix Block Comparison
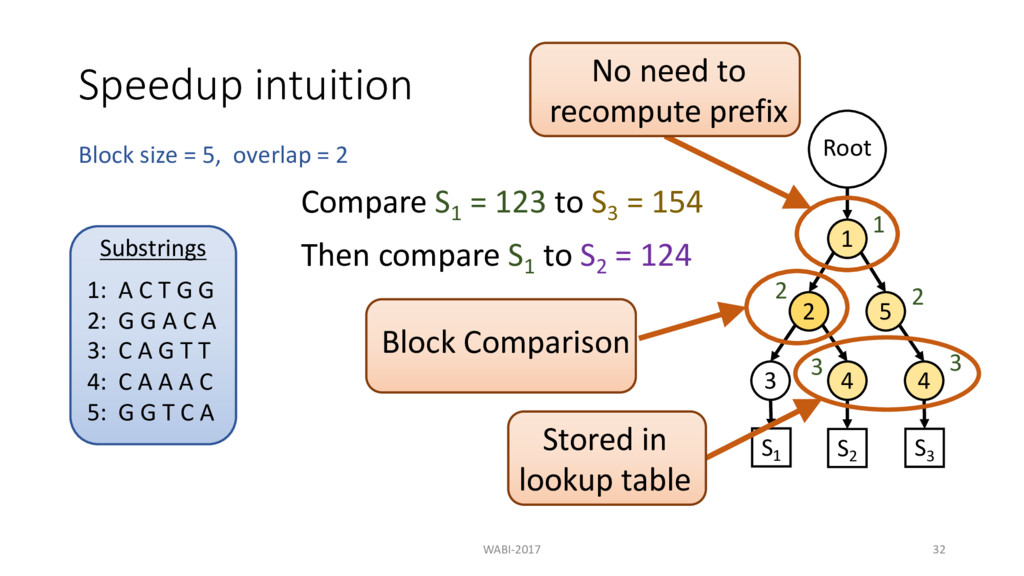
2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C 5: G G T C A Root 1 2 3 4 5 4 S1 S2 S3 Compare S1 = 123 to S3 = 154 Then compare S1 to S2 = 124 1 2 3 2 3 No need to recompute prefix Block Comparison
2 Substrings 1: A C T G G 2: G G A C A 3: C A G T T 4: C A A A C 5: G G T C A Root 1 2 3 4 5 4 S1 S2 S3 Compare S1 = 123 to S3 = 154 Then compare S1 to S2 = 124 1 2 3 2 3 Stored in lookup table No need to recompute prefix Block Comparison
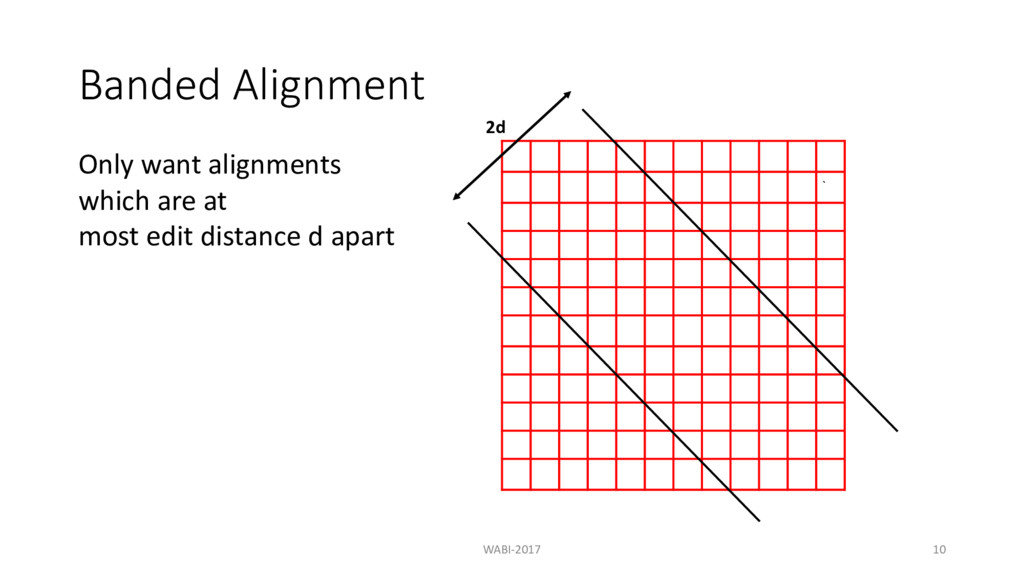
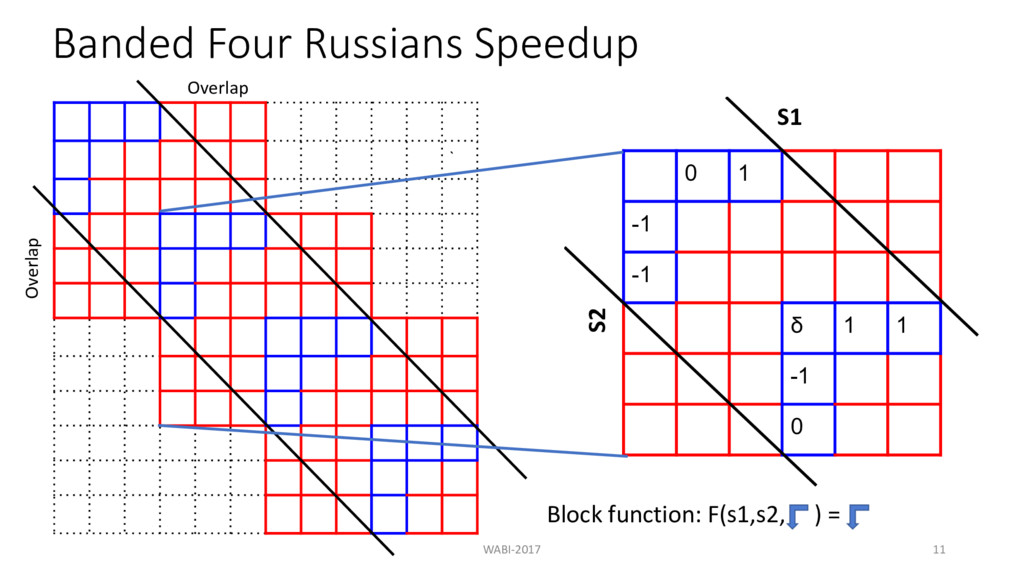
at each level in the tree d = maximum allowed edit distance m = length of the sequence n = number of sequences Banded alignment: O(n2md) Theorem: If = ≤ ? @A B , all pairwise edit distance can be computed in O(n2m) time. WABI-2017 37
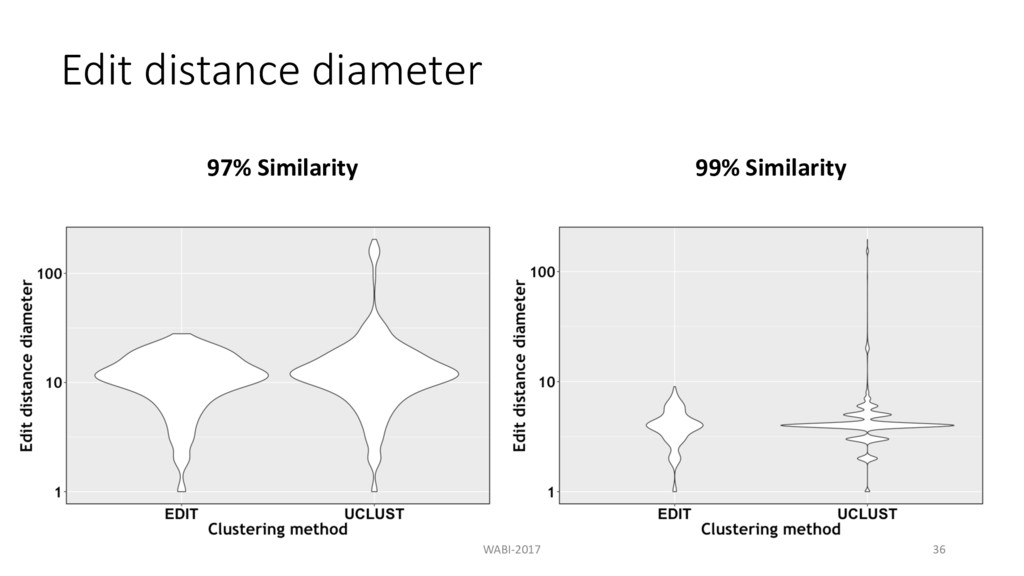
prefix • Combined 4-Russian’s method with banded alignment • Find compact and accurate clusters compared to UCLUST at high similarity • Preprocessing step for complicated clustering methods WABI-2017 38
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![WABI-2017 40 Thank You Questions? Contact: [email protected] and [email protected]](https://files.speakerdeck.com/presentations/5d2541d26df340ed8ebaa19ff3f69088/slide_39.jpg){kind=link}