It constitutes of basic introduction of regulatory affairs in pharmaceuticals, career in pharma regulatory affairs, job opportunities and future aspect. GIBTIndia offers job oriented e- learning courses. Kindly visit us at www.gibtindia.com
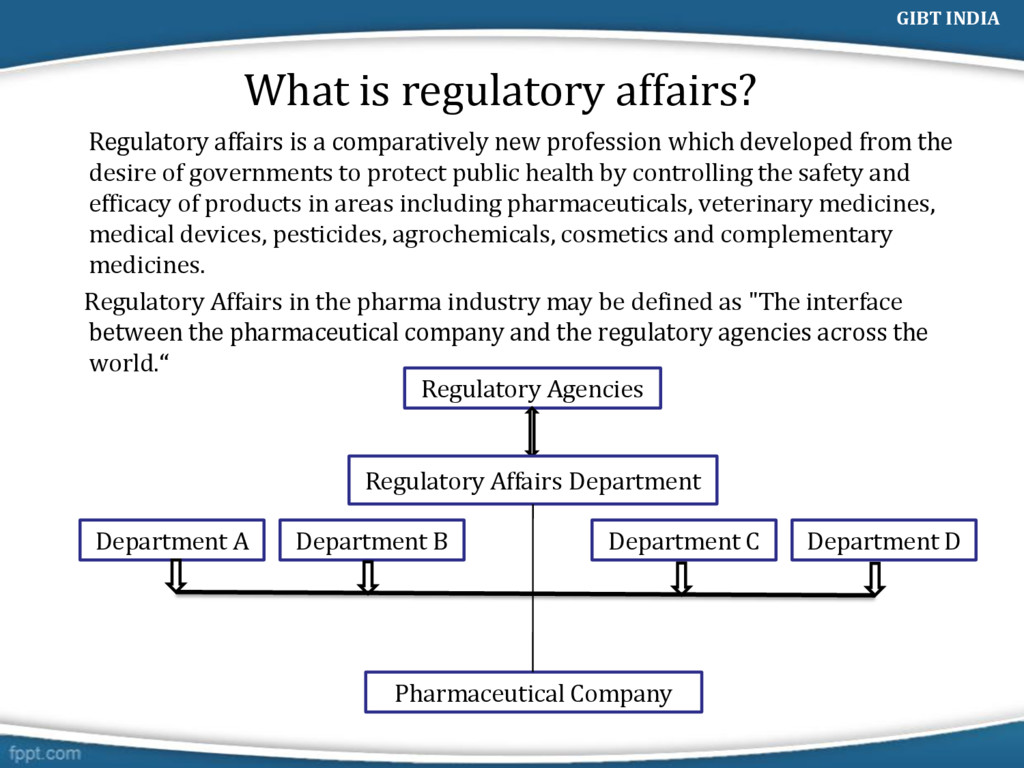
profession which developed from the desire of governments to protect public health by controlling the safety and efficacy of products in areas including pharmaceuticals, veterinary medicines, medical devices, pesticides, agrochemicals, cosmetics and complementary medicines. Regulatory Affairs in the pharma industry may be defined as "The interface between the pharmaceutical company and the regulatory agencies across the world.“ Regulatory Agencies Regulatory Affairs Department Department A Department B Department C Department D Pharmaceutical Company GIBT INDIA
changing legislation Registration documents to regulatory agencies To give strategic and technical advice to R&D, Production, QC Department etc. GIBT INDIA
Pharmaceutical industry is the most regulated of all the industries. Regulations are put in order to develop the most efficient and safe pharmaceutical products. It takes more than 8 to 15years to develop a new drug product & costs more than $800 million. Regulatory affairs provides insight / guidance into this development through agency wisdom collected in guidance, previous experience, market precedence, etc. and hence helps to reduce the number of development failures. GIBT INDIA
(across the world) CDSCO- India USFDA- United States EMEA- European countries MHRA- United Kingdom TGA- Australia MCC- South Africa Internal Departments of Organizations Product Developments Clinical Research & Trials Licensing Manufacturing Quality Assurance & Control Marketing GIBT INDIA
documents that contain all the technical data of pharmaceutical product to be approved / registered / marketed in a country. It is most commonly called as Registration Dossier, In US: New Drug Application (NDA) In EU: Marketing Authorization Appliation (MAA) GIBT INDIA
sites, Facilities, Operating Procedures and Personnel (no longer applicable) Type II- DS, Intermediate and material used in their preparation or drug product Type III- Packaging Material Type IV- Excipient, Colorant, Flavor or Material used in their preparation Type V- FDA Accepted Reference Information (FDA discourages its use) US United State Drug Master File (US- DMF) EU European Drug Master File (EDMF) or Active Substance Master File (ASMF) GIBT INDIA
is an format set by ICH which was agreed by Regulatory Agencies of Europe, Japan and the US. Its electronic version called as electronic Common Technical Document (eCTD) GIBT INDIA
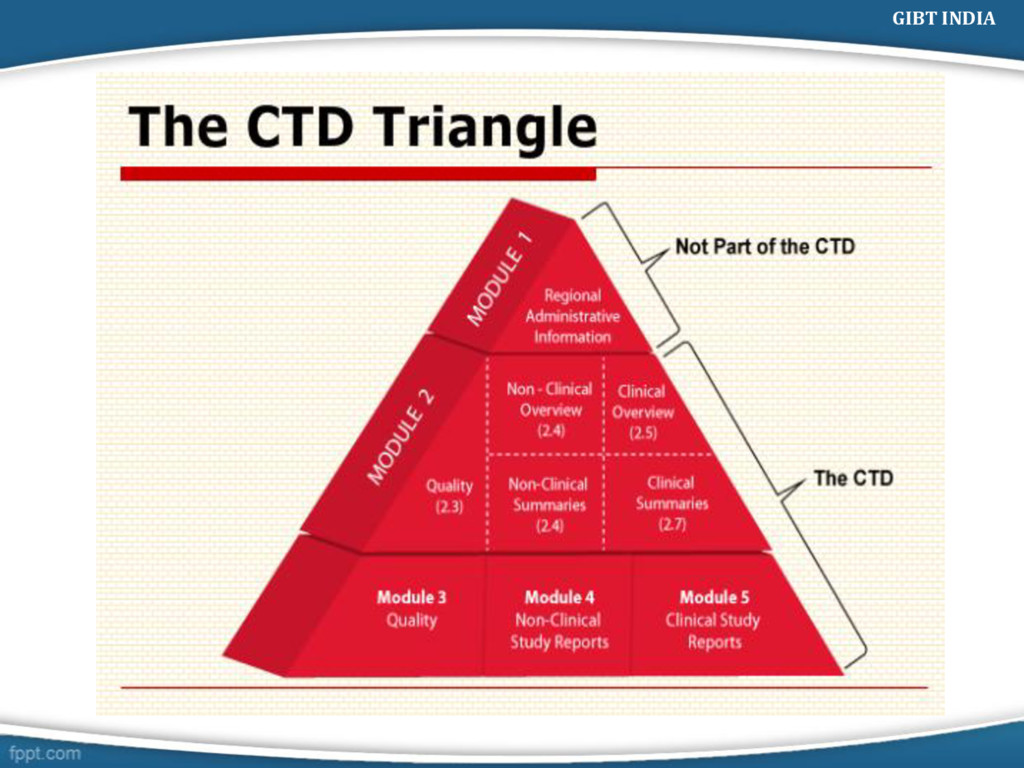
Debarment Certificate Letter of Authorization (LOA)/DMF Letter Labeling Text For EU Application form Summary of product characteristics Labelling text andmock-ups Information about the experts Environmental riskassesment Description of the pharmacovigilance system Risk management plan Module 1 Administrative Information(Region Specific) Should contain documents specific to each region GIBT INDIA
in the following order: CTD DOC (Module 2- 5) CD Introduction Quality Overall Summary Non- Clinical Overview Clinical Overview Non Clinical Summary Clinical Summary GIBT INDIA
Tabular listing of Clinical Studies Clinical study reports a) Reports of Biopharmaceutical (BA-BE) Study b) Reports of Pharmacokinetic (biomaterial) study c) Reports of Pharmacokinetic (PK) Studies d) Reports of Pharmacodynamic (PD) Studies e) Reports of Efficacy and Safety studies f) Reports of Post – Marketing experience g) Case Report Forms & Individual patient listings Literature References GIBT INDIA
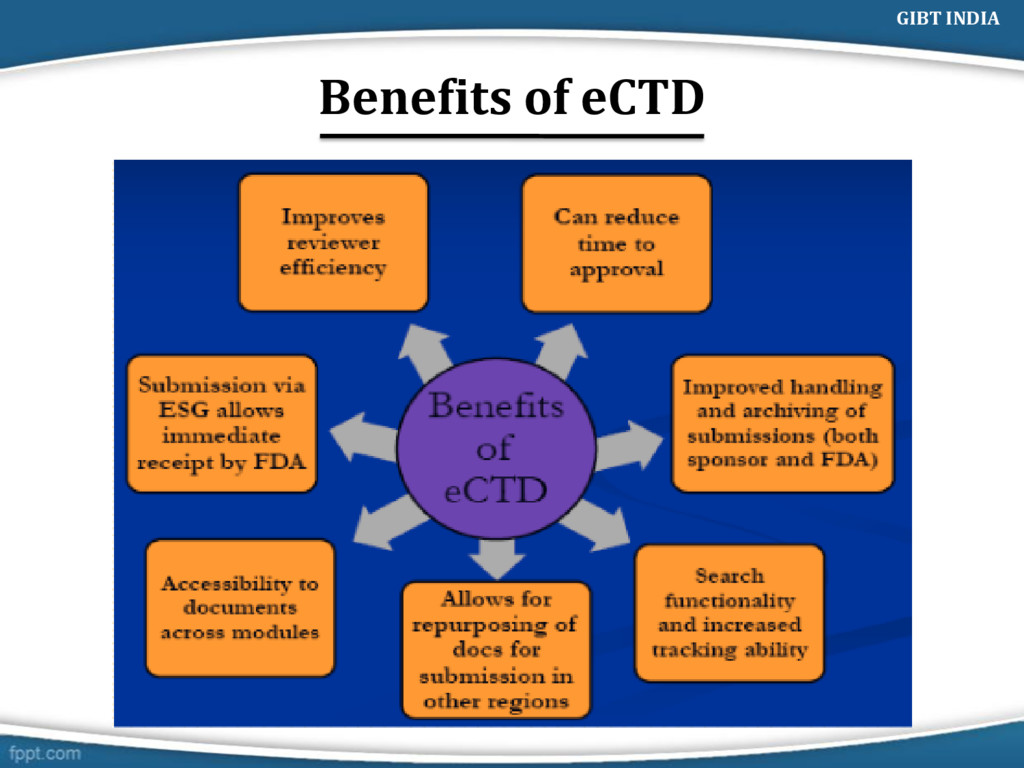
Common Technical Document (eCTD). eCTD composed of two types of specification a) Content Specification- As defined by ICH b) Technical Specification- Electronic software’s CTDTOC (PDF) (Paper) eCTD XML Backbone GIBT INDIA
options PDF documents linked via XML backbone Increased document granularity Transparency of entire submission Ease of navigation and review GIBT INDIA
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}