A recap of recent work from our R Bioconductor-powered Team Data Science group at the Lieber Institute for Data Science.
Team website: https://lcolladotor.github.io/
Topics Presented & Presenters:
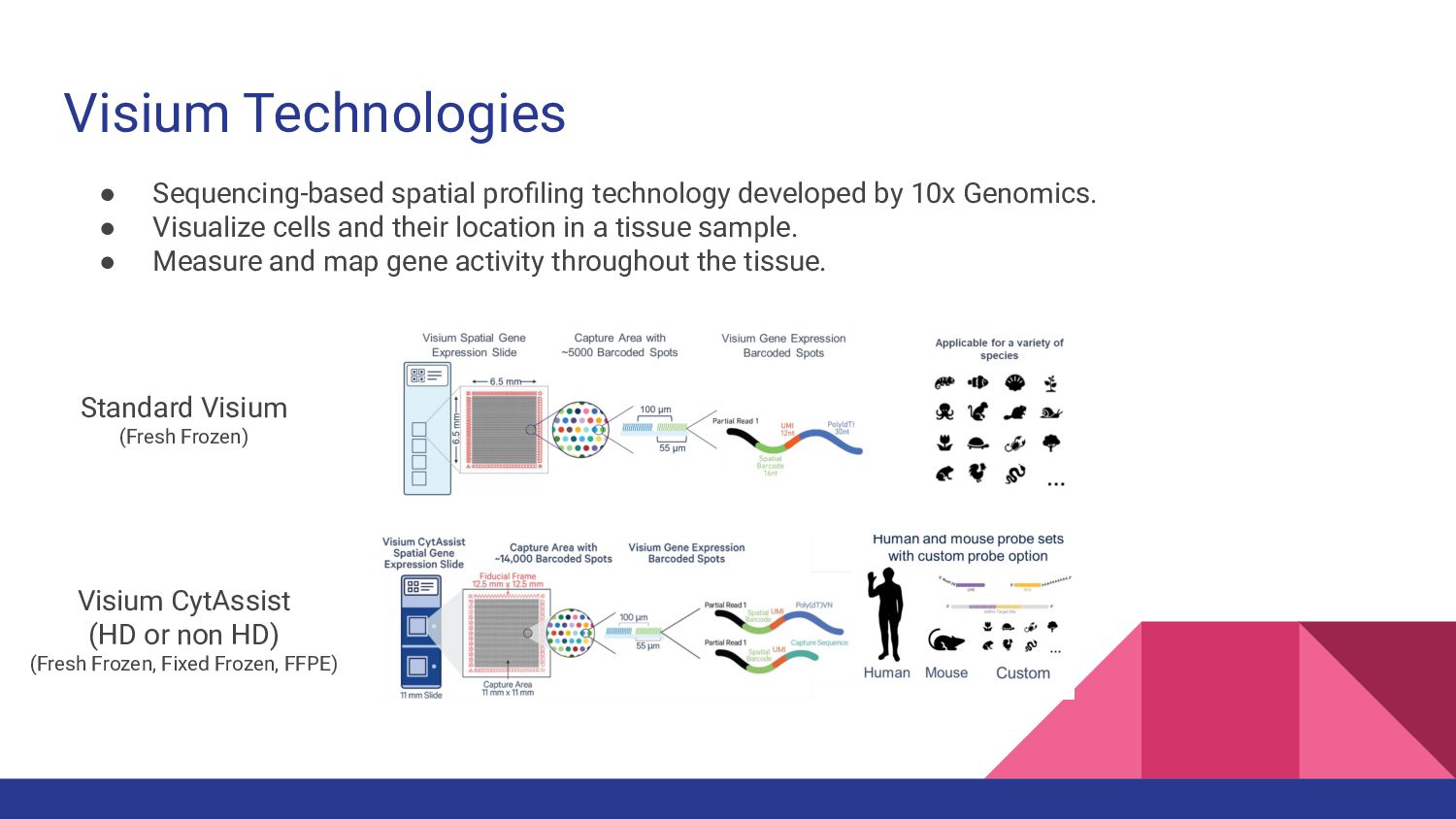
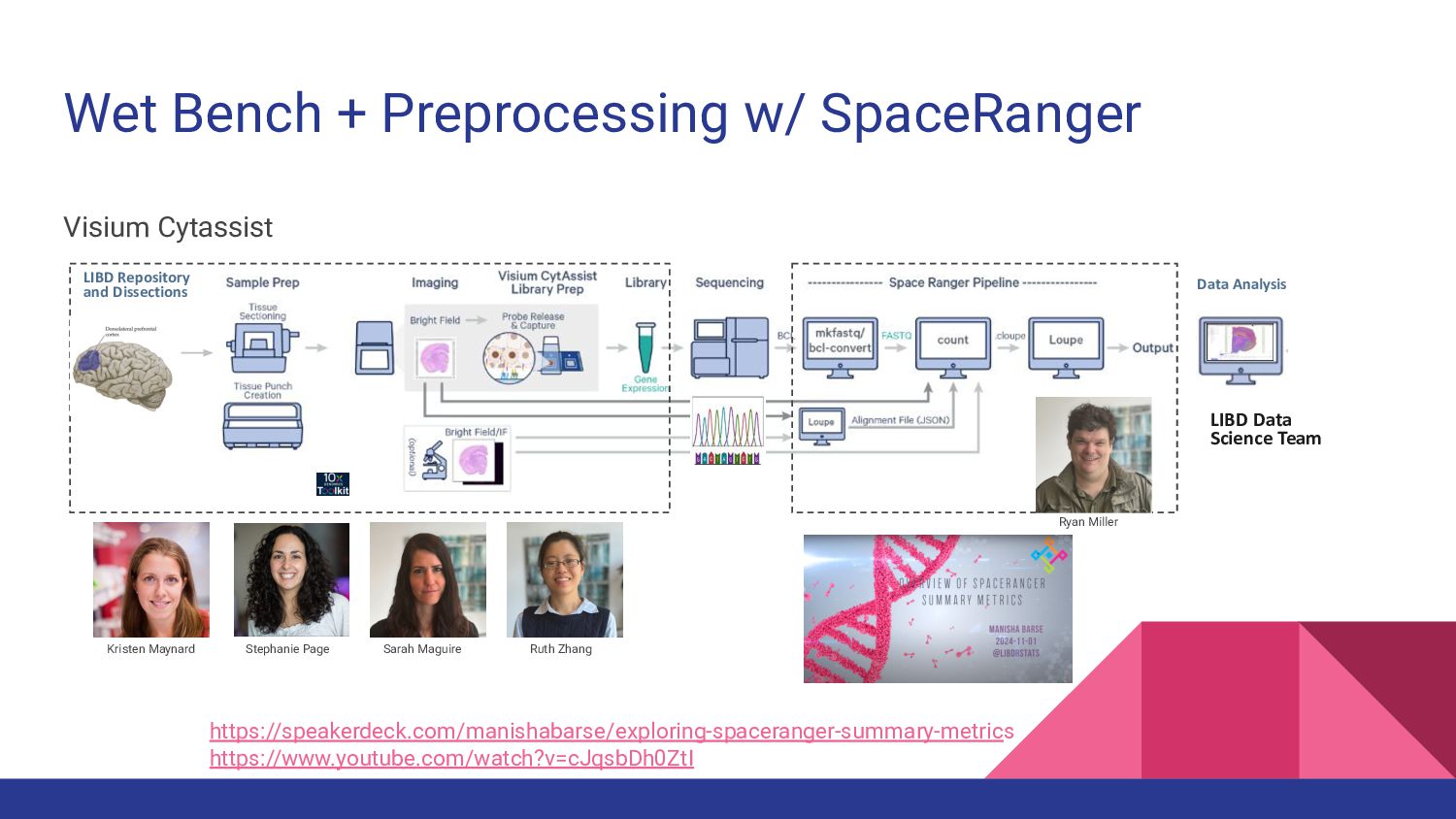
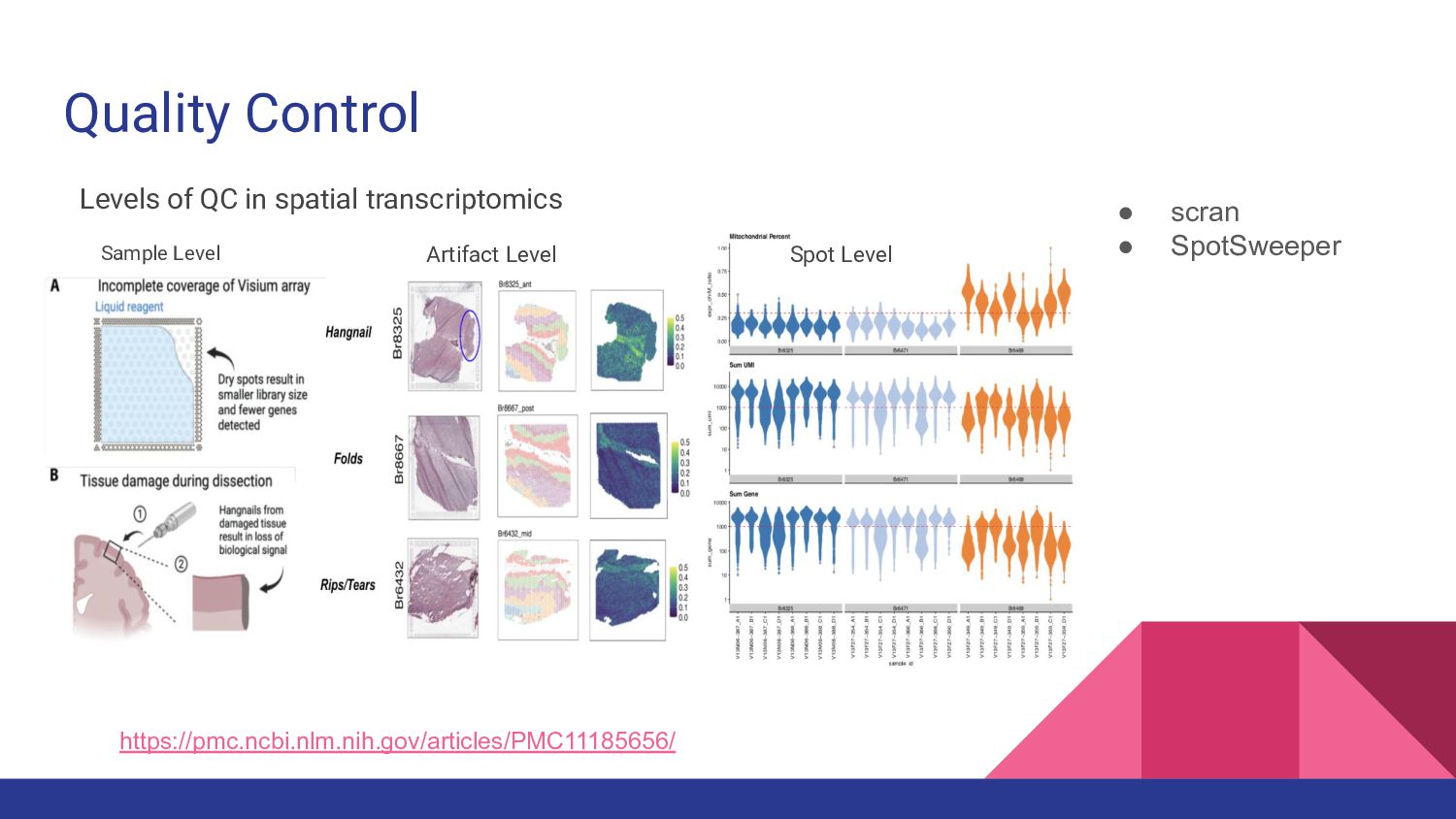
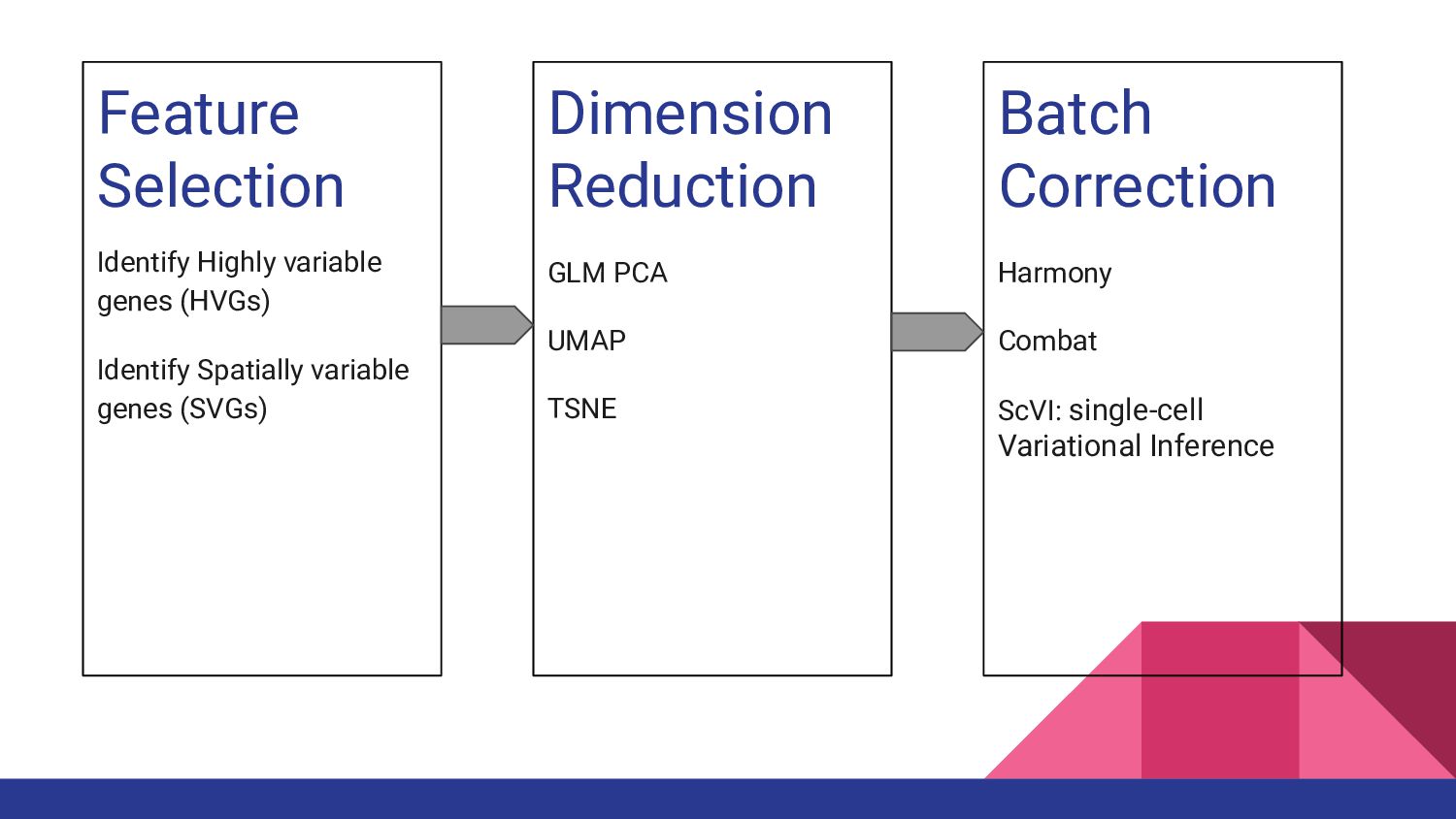
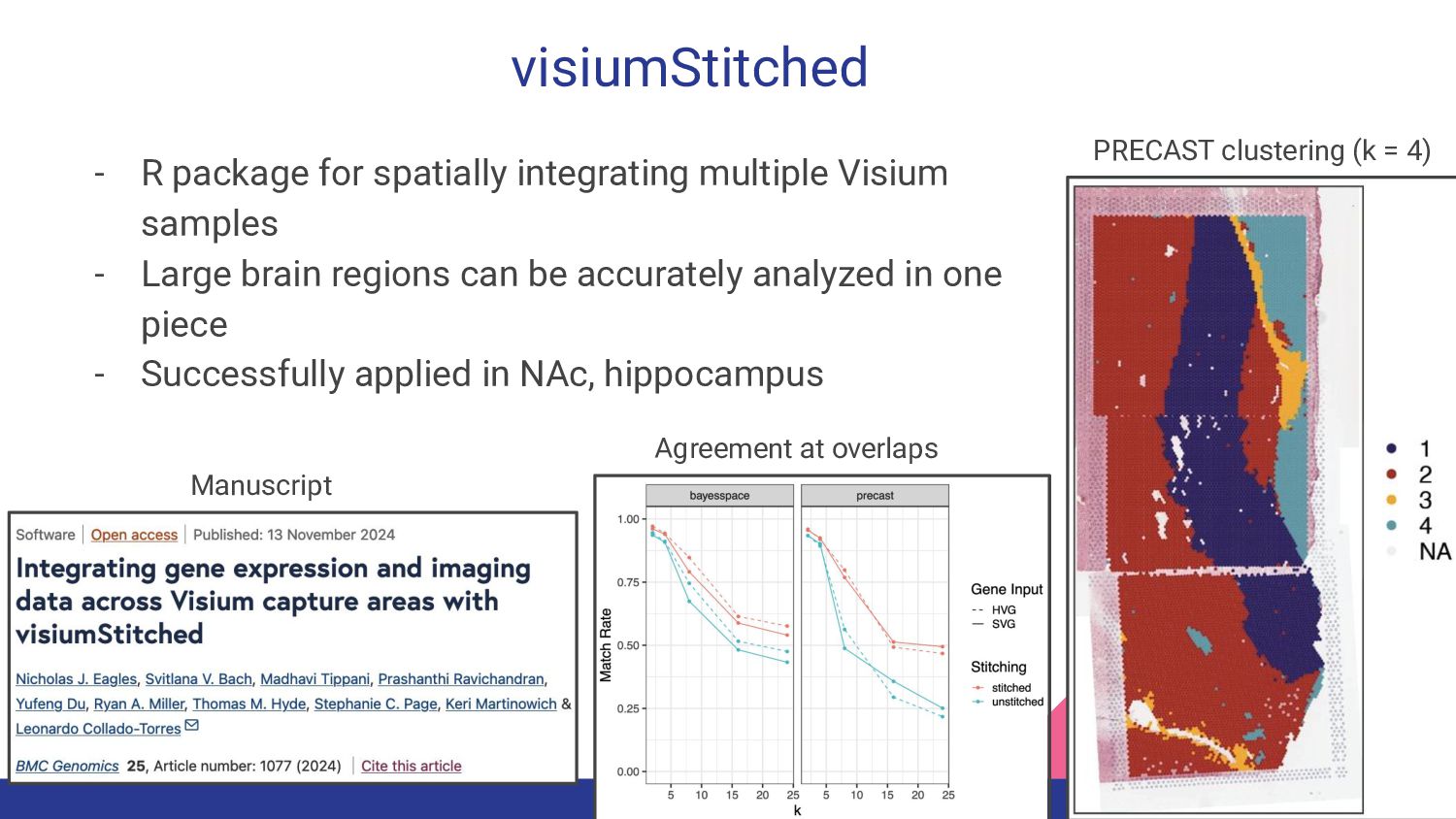
1. Spatially Resolved Transcriptomics with Visium - Manisha Barse
2. Visium HD data analysis - Nick Eagles
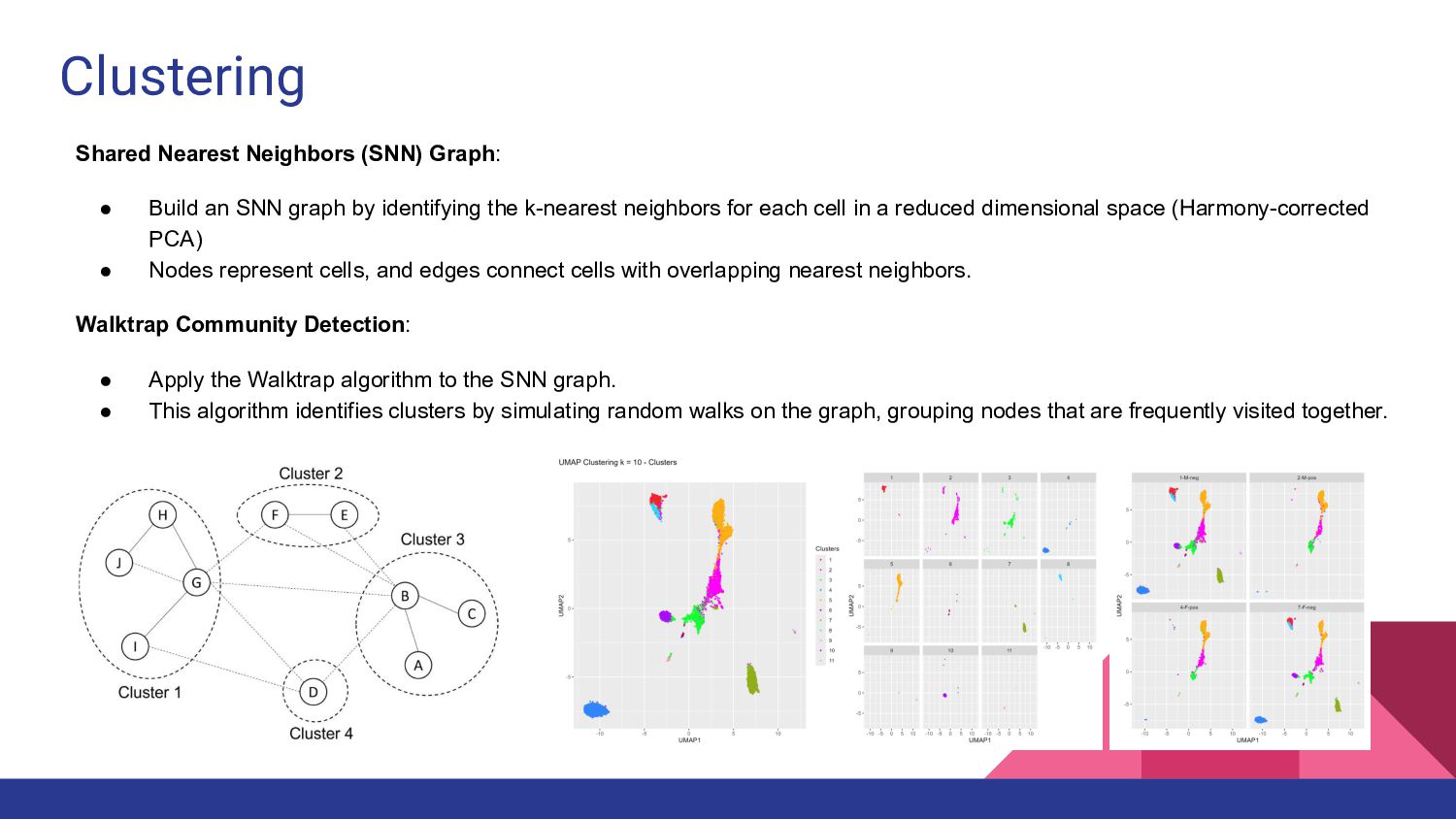
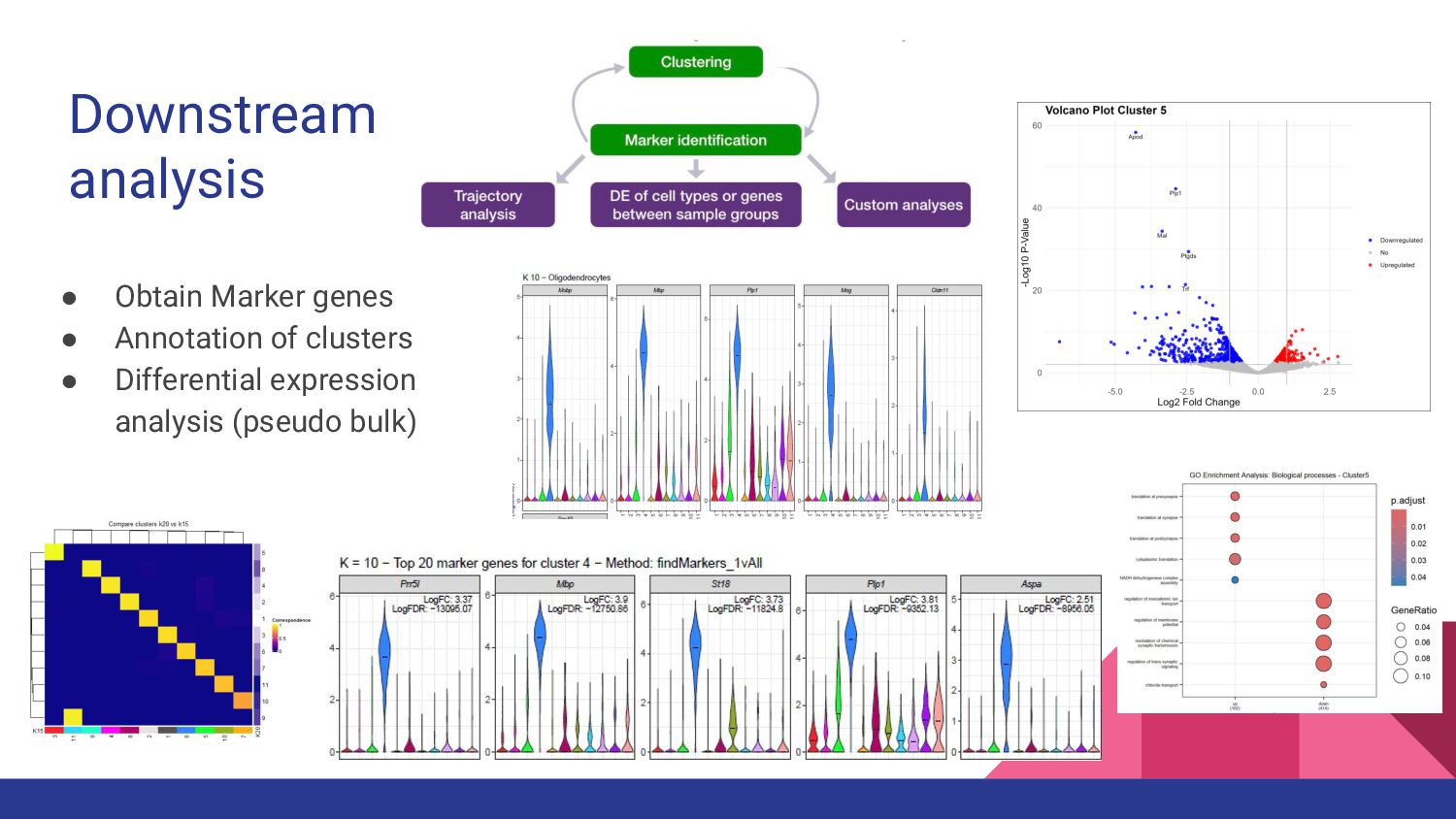
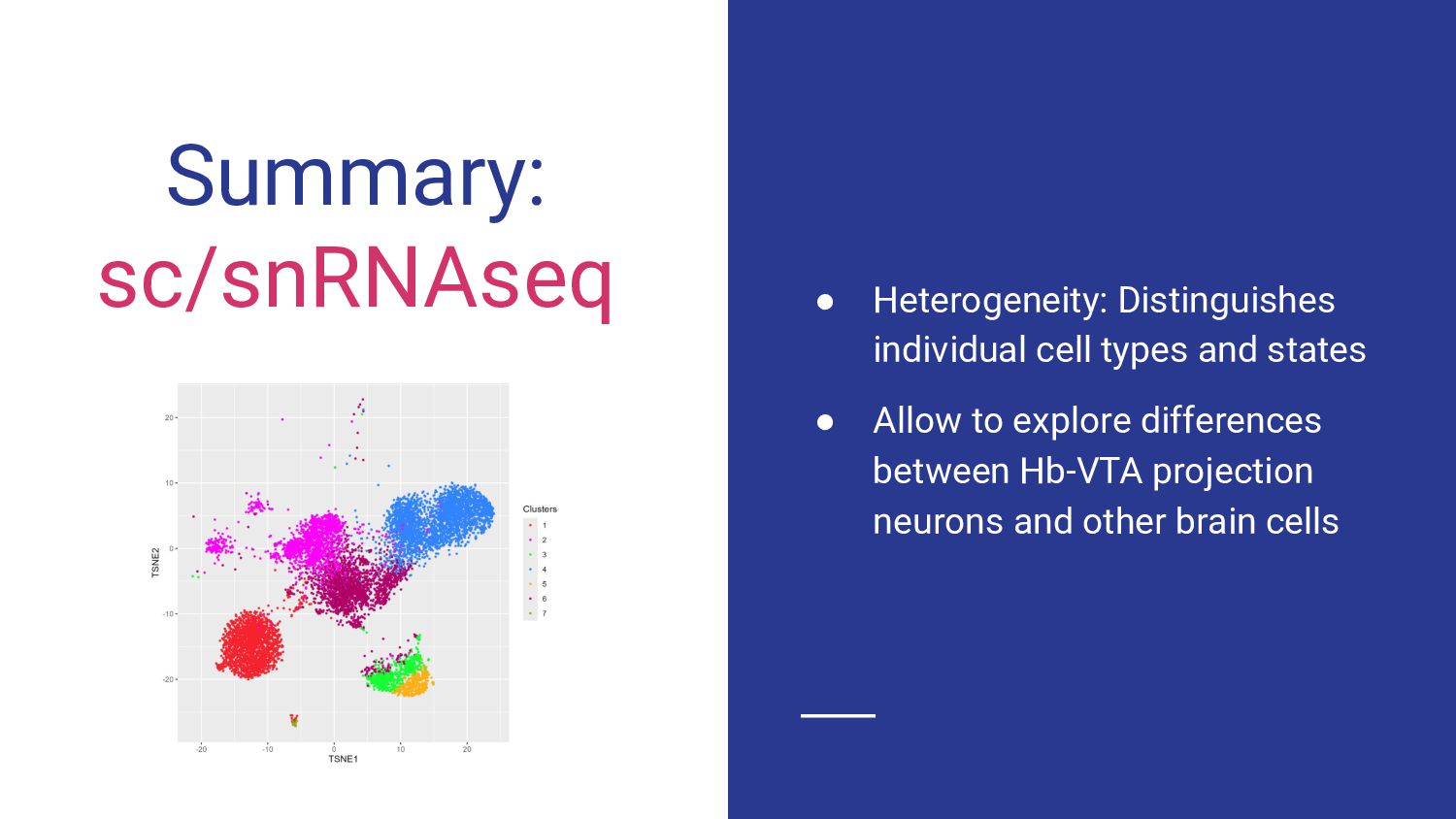
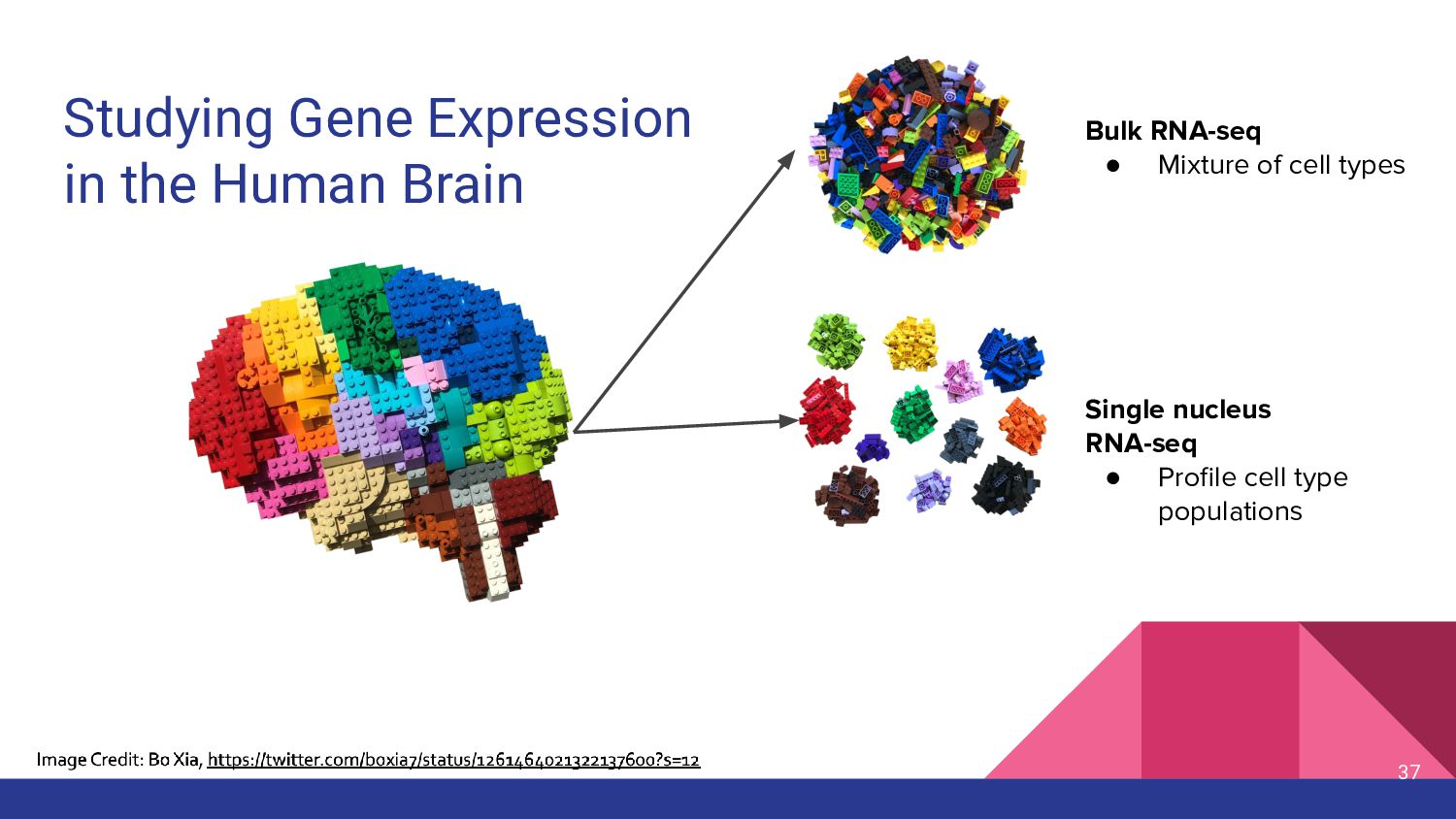
3. sc/snRNA-seq data analysis - Melissa Mayén Quiroz
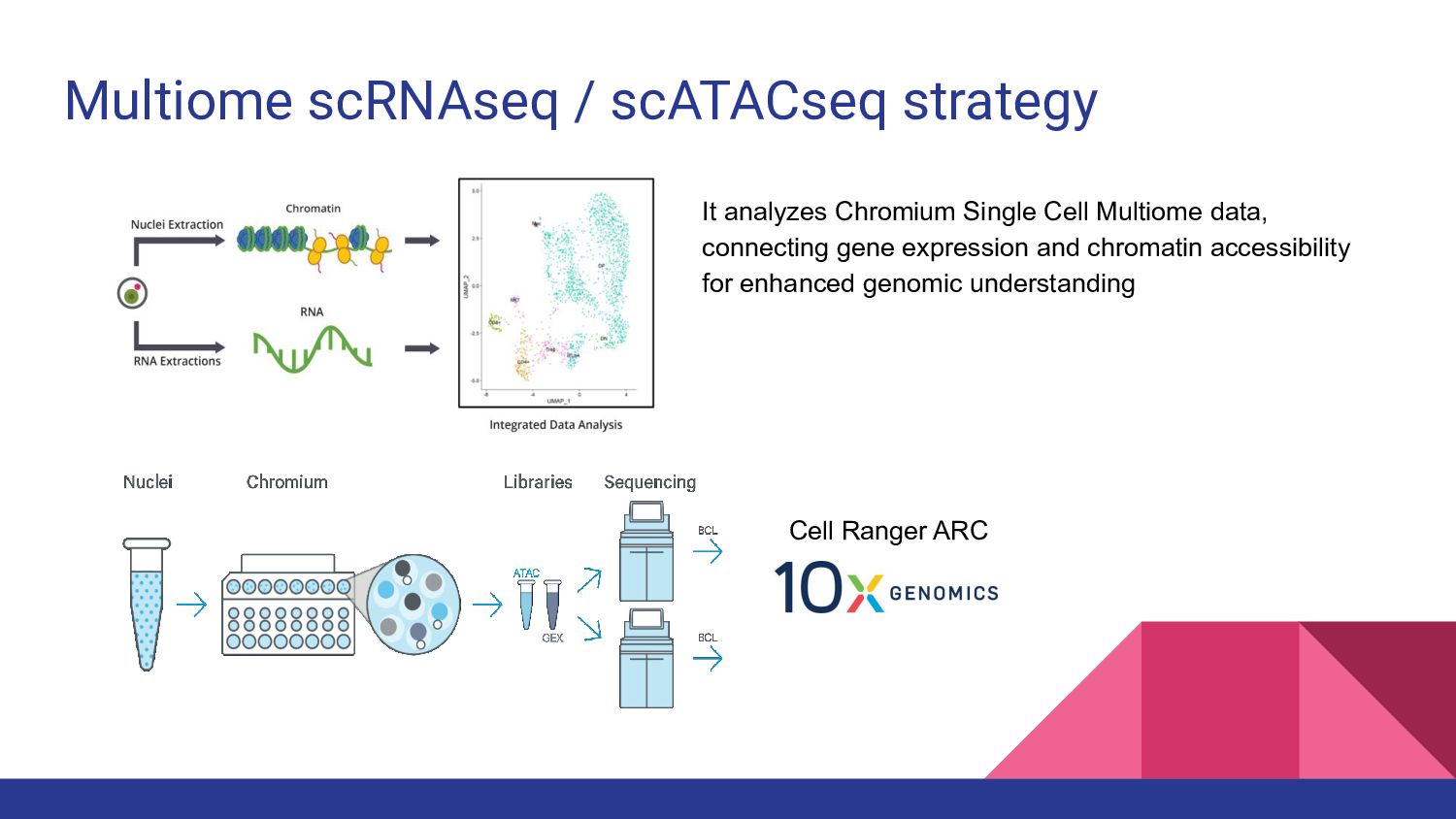
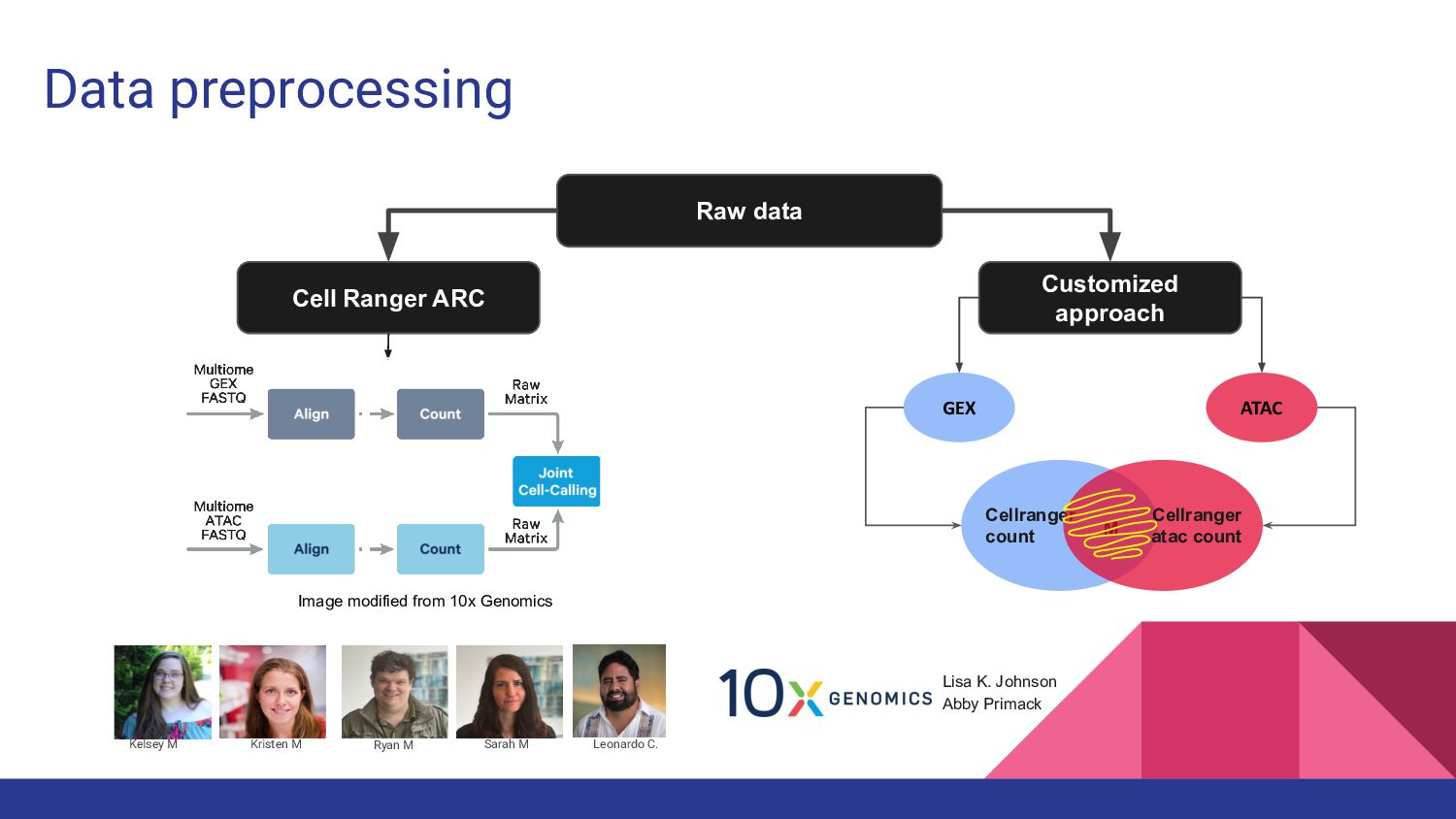
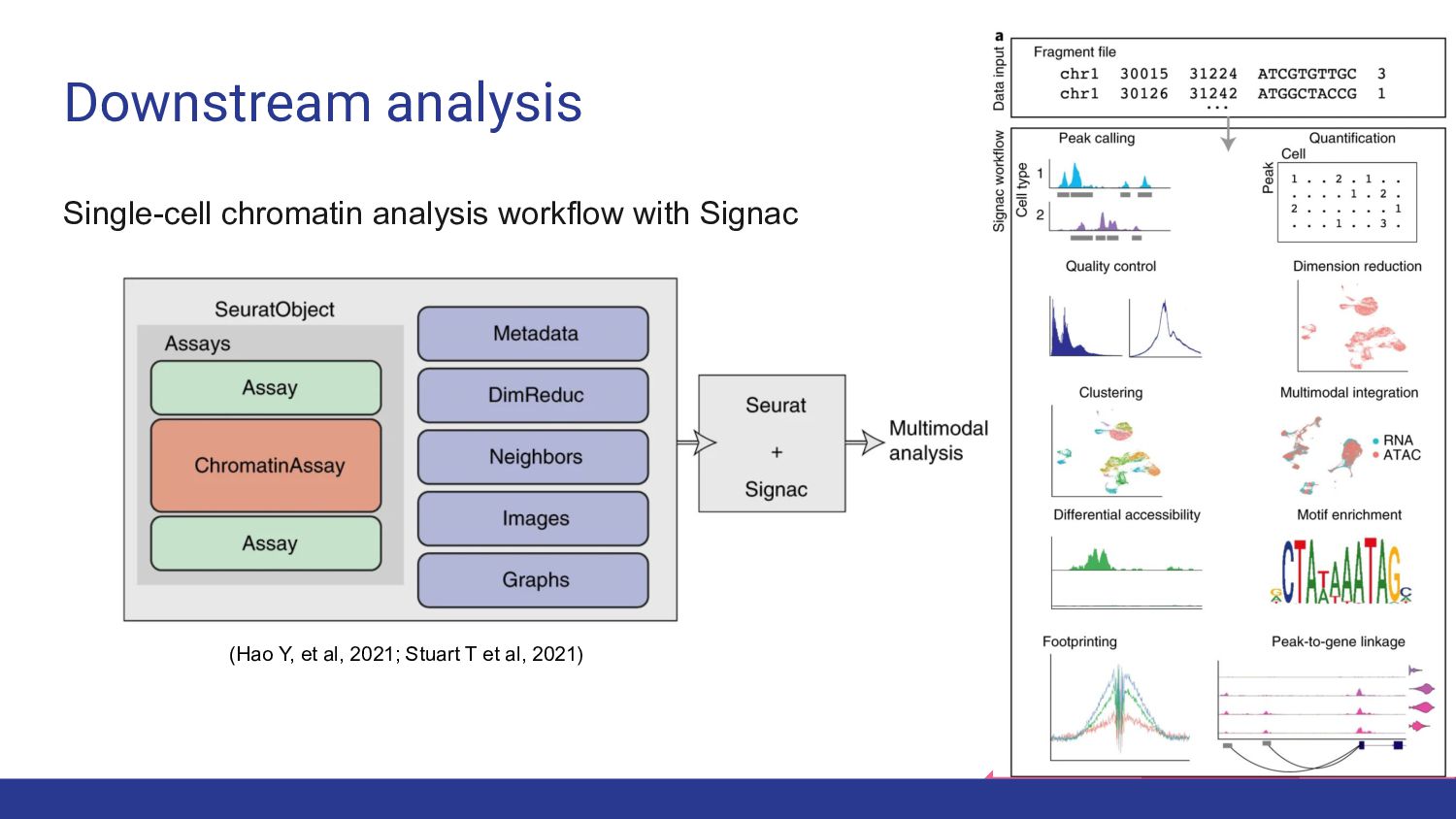
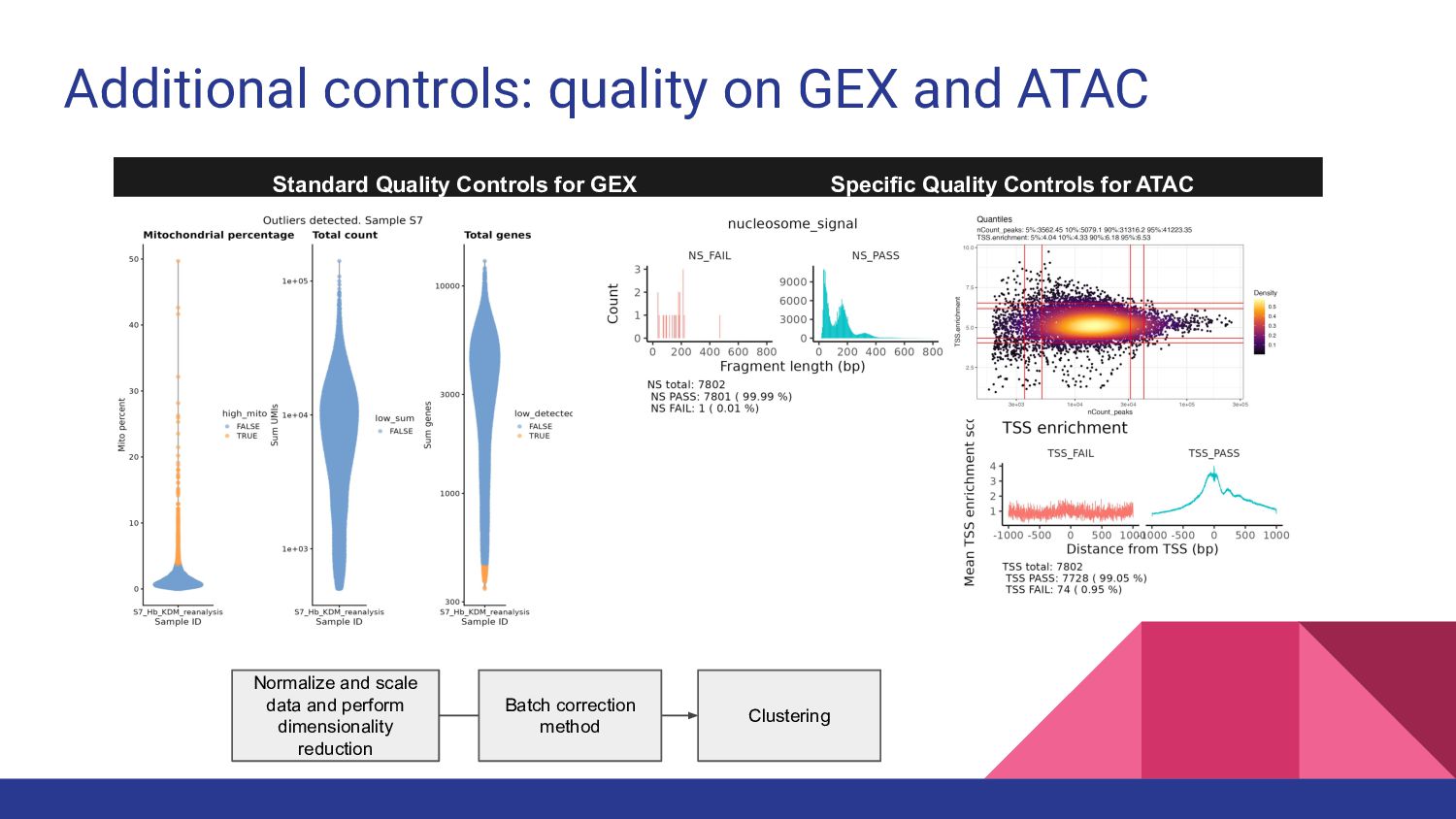
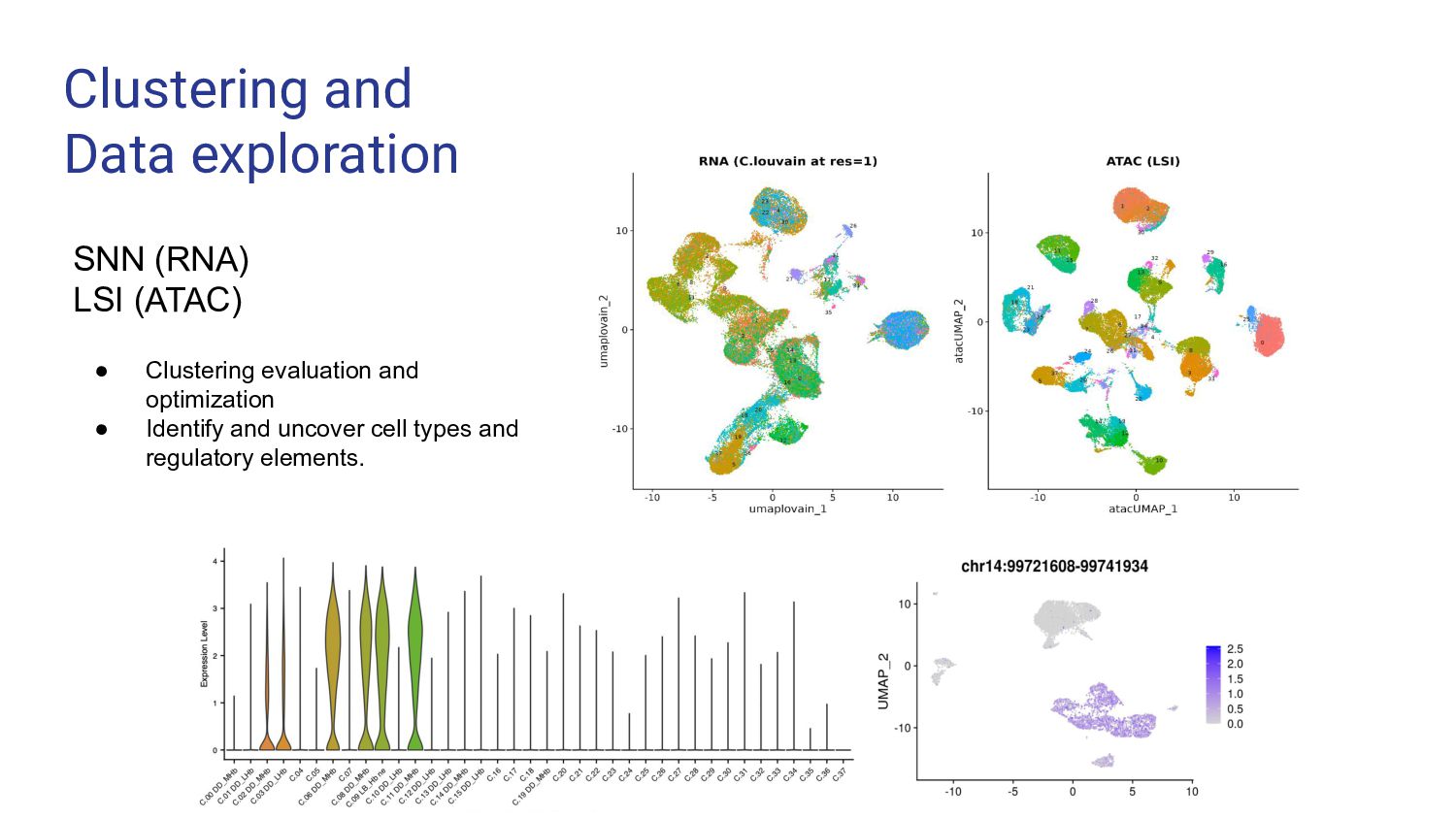
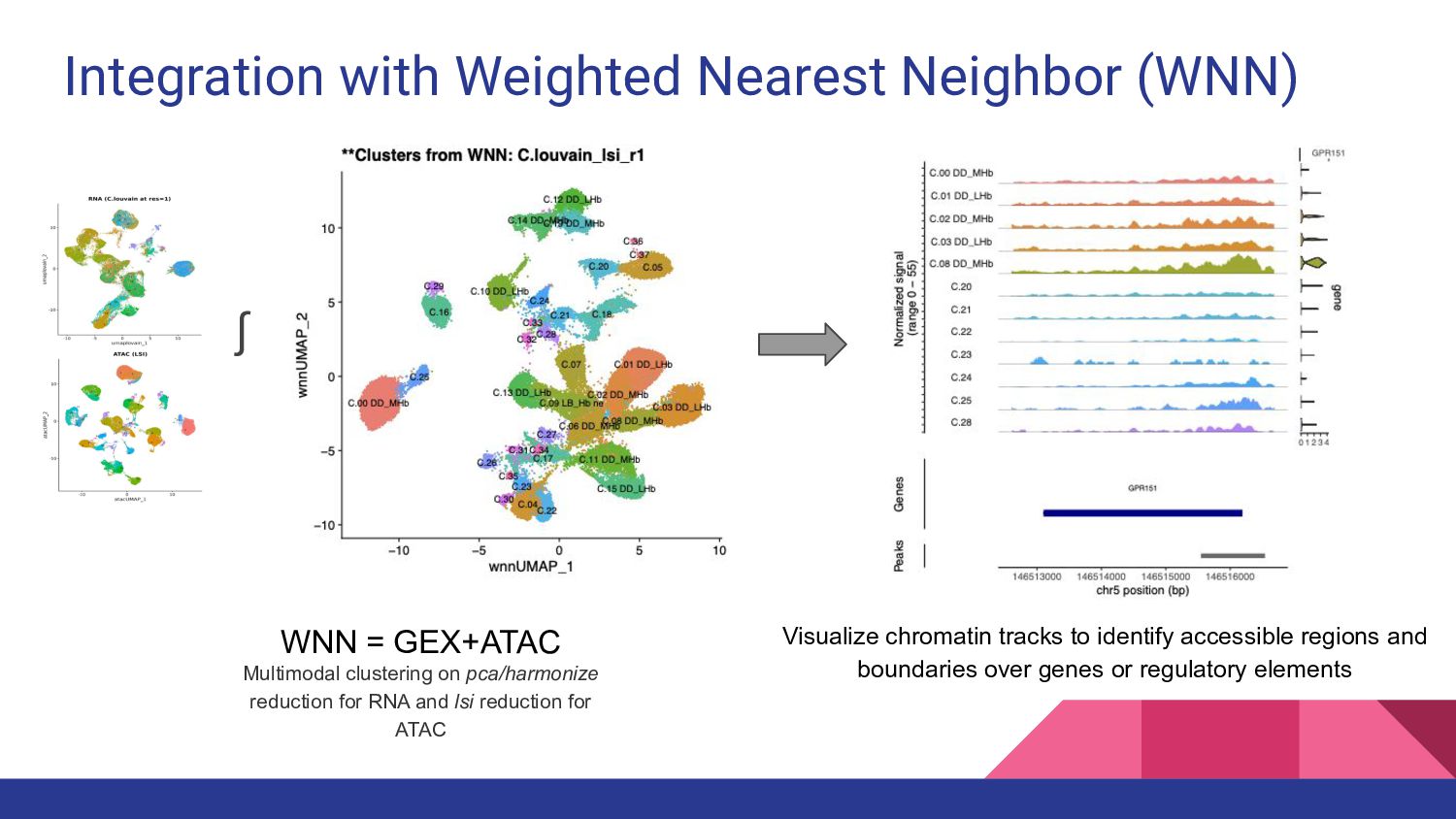
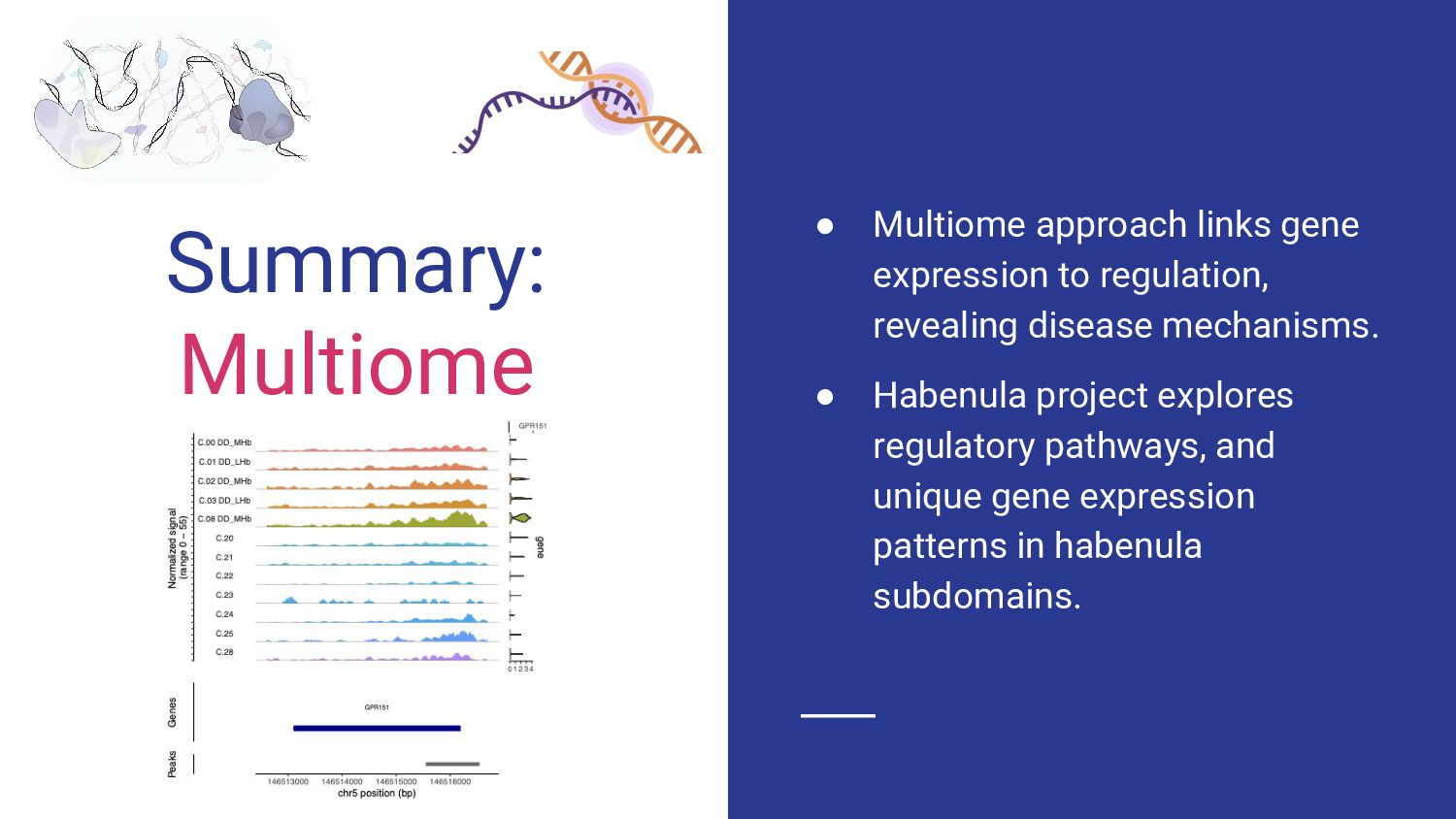
4. Multi-ome data analysis - Cynthia Soto Cardinault
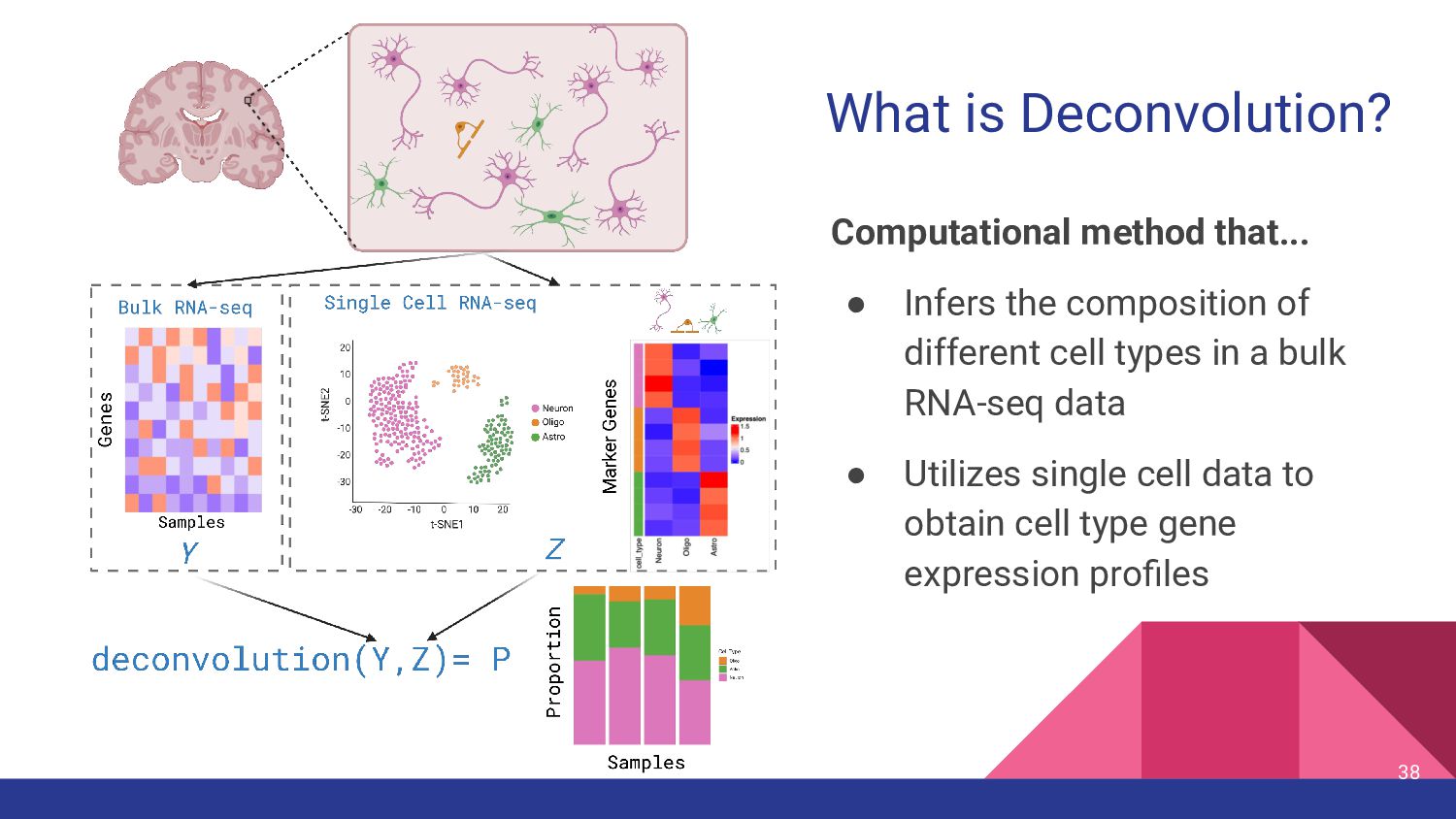
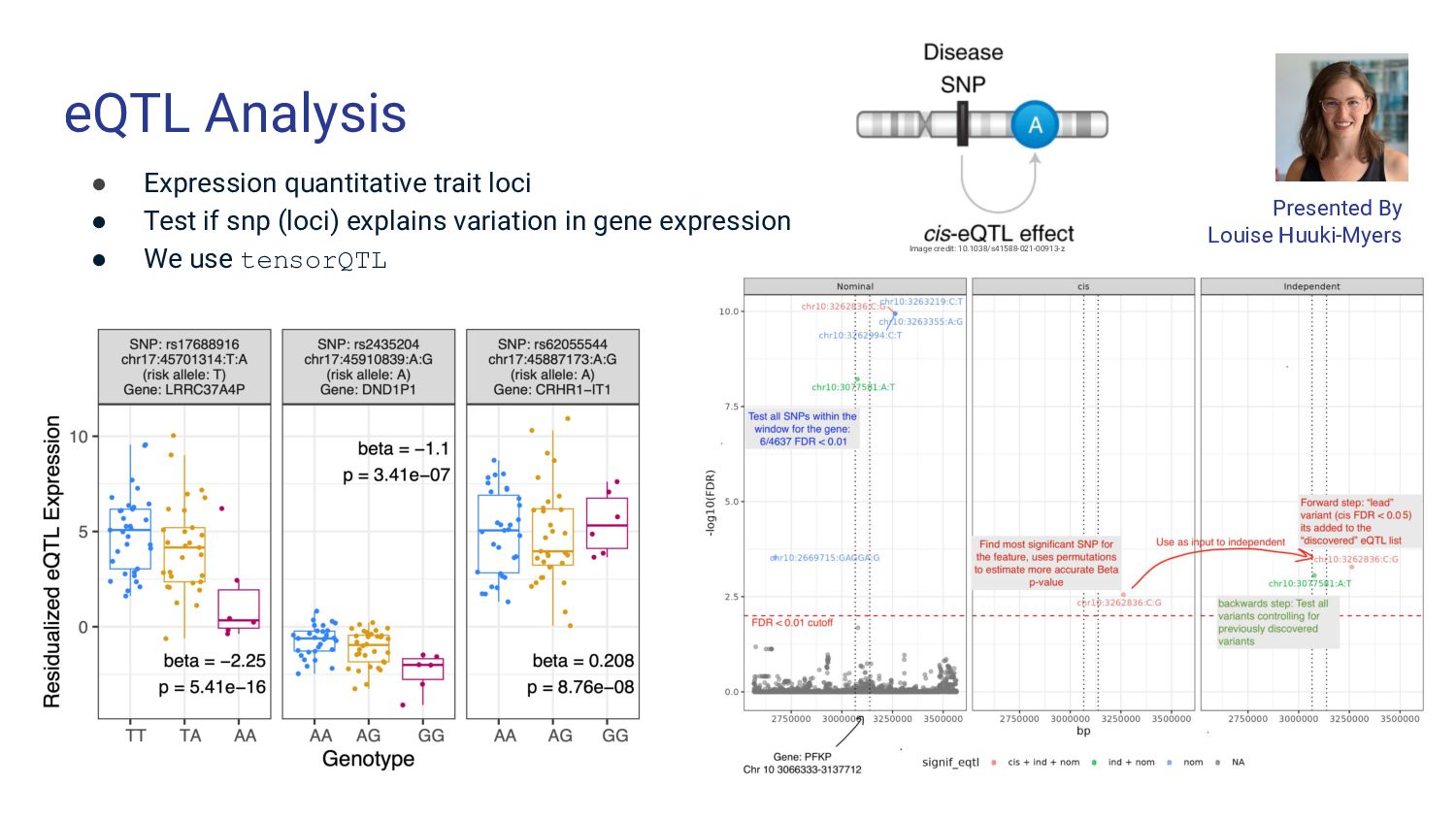
5. Bulk RNA-seq deconvolution - Louise Huuki-Myers
6. Bulk RNA-seq data analysis - Louise Huuki-Myers & Nick Eagles
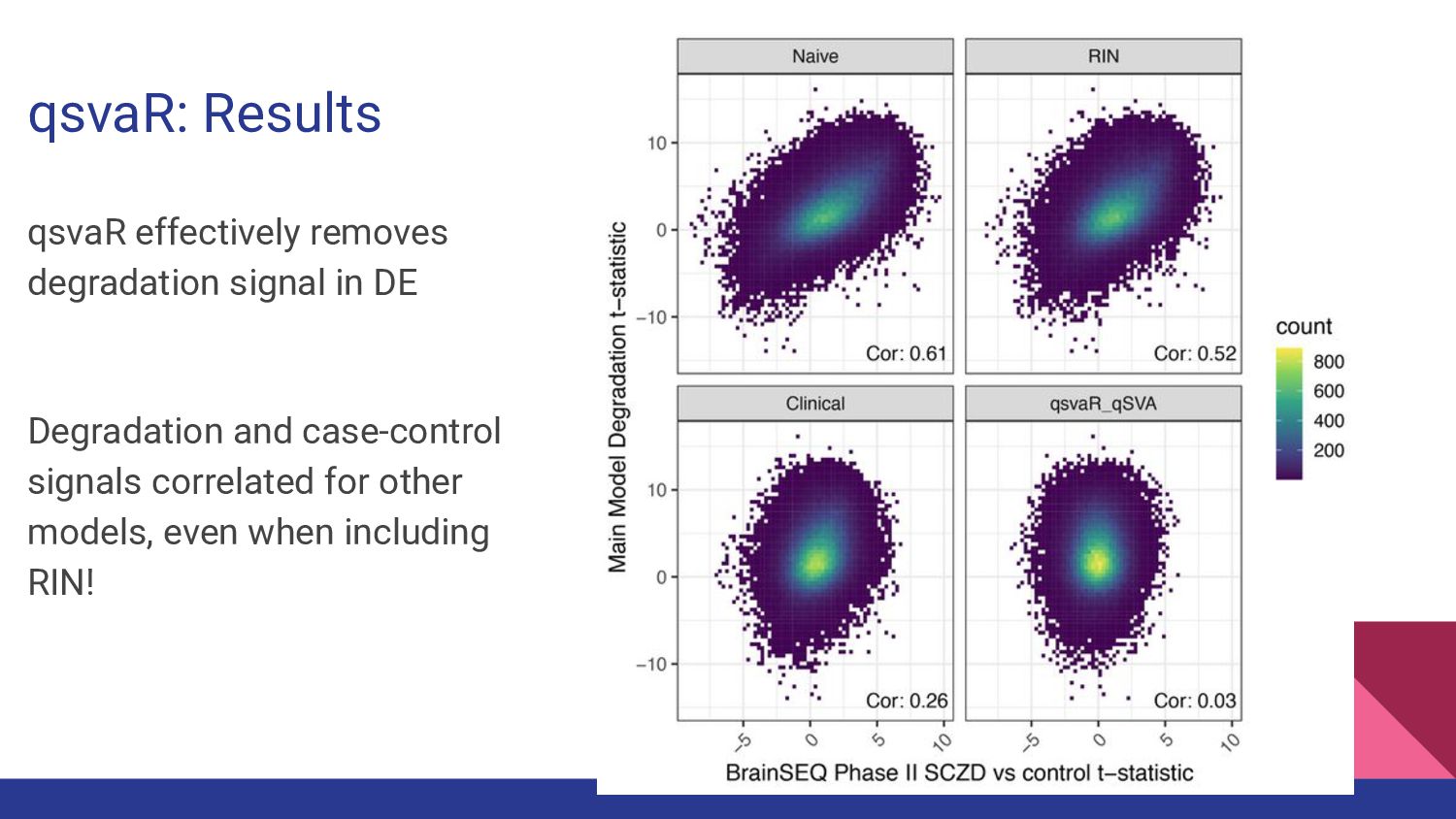
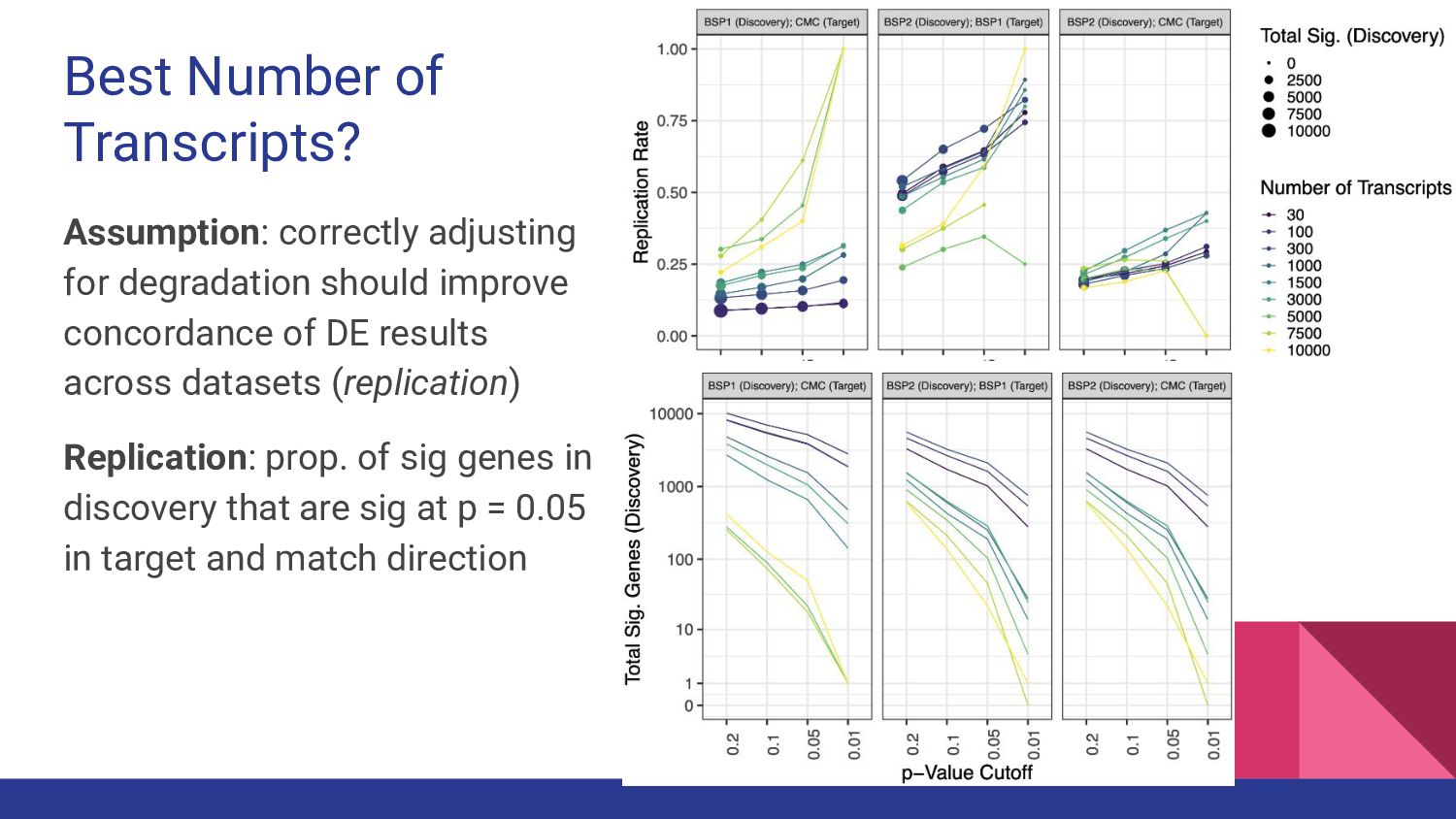
7. qsvaR - Nick Eagles
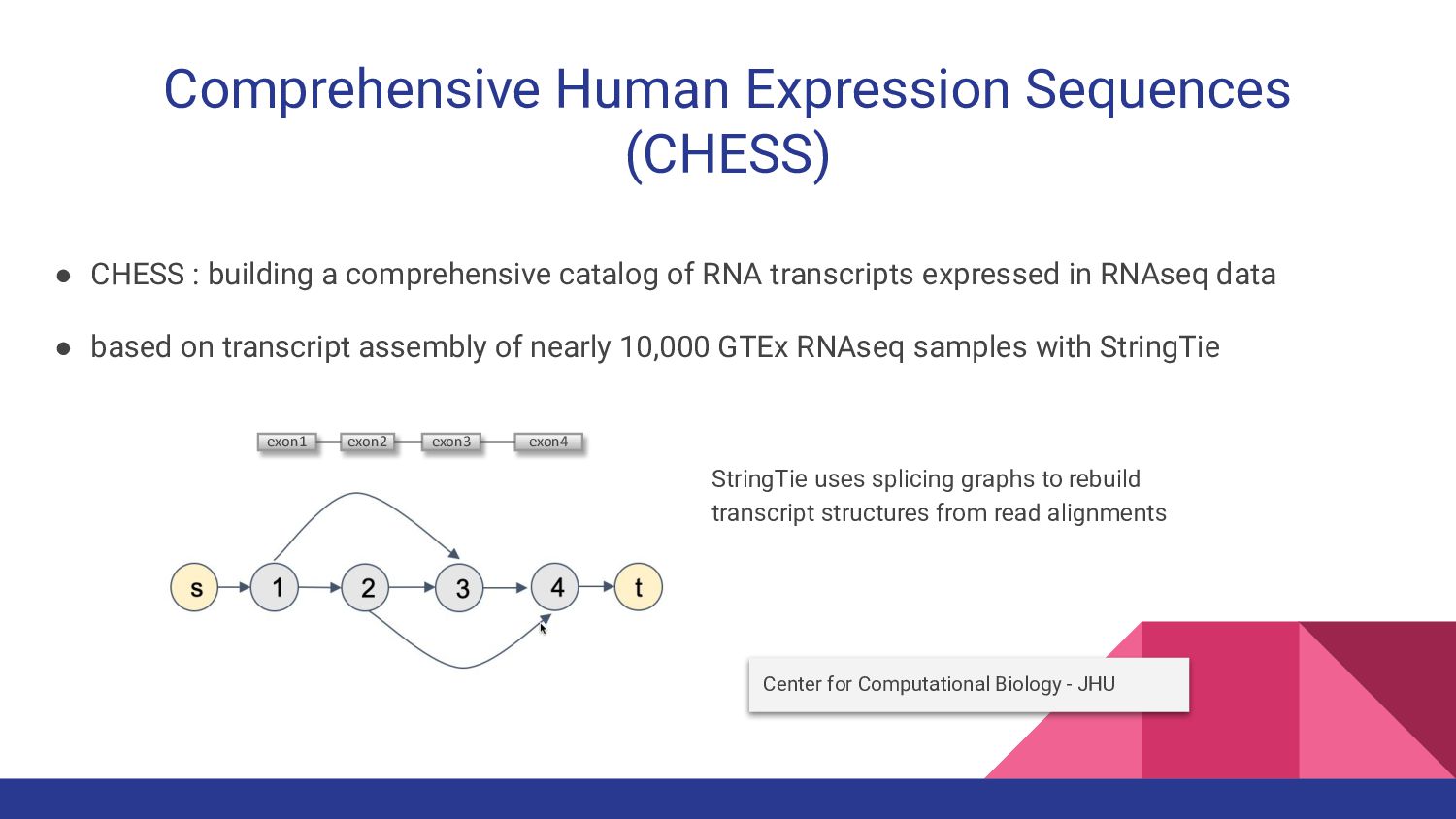
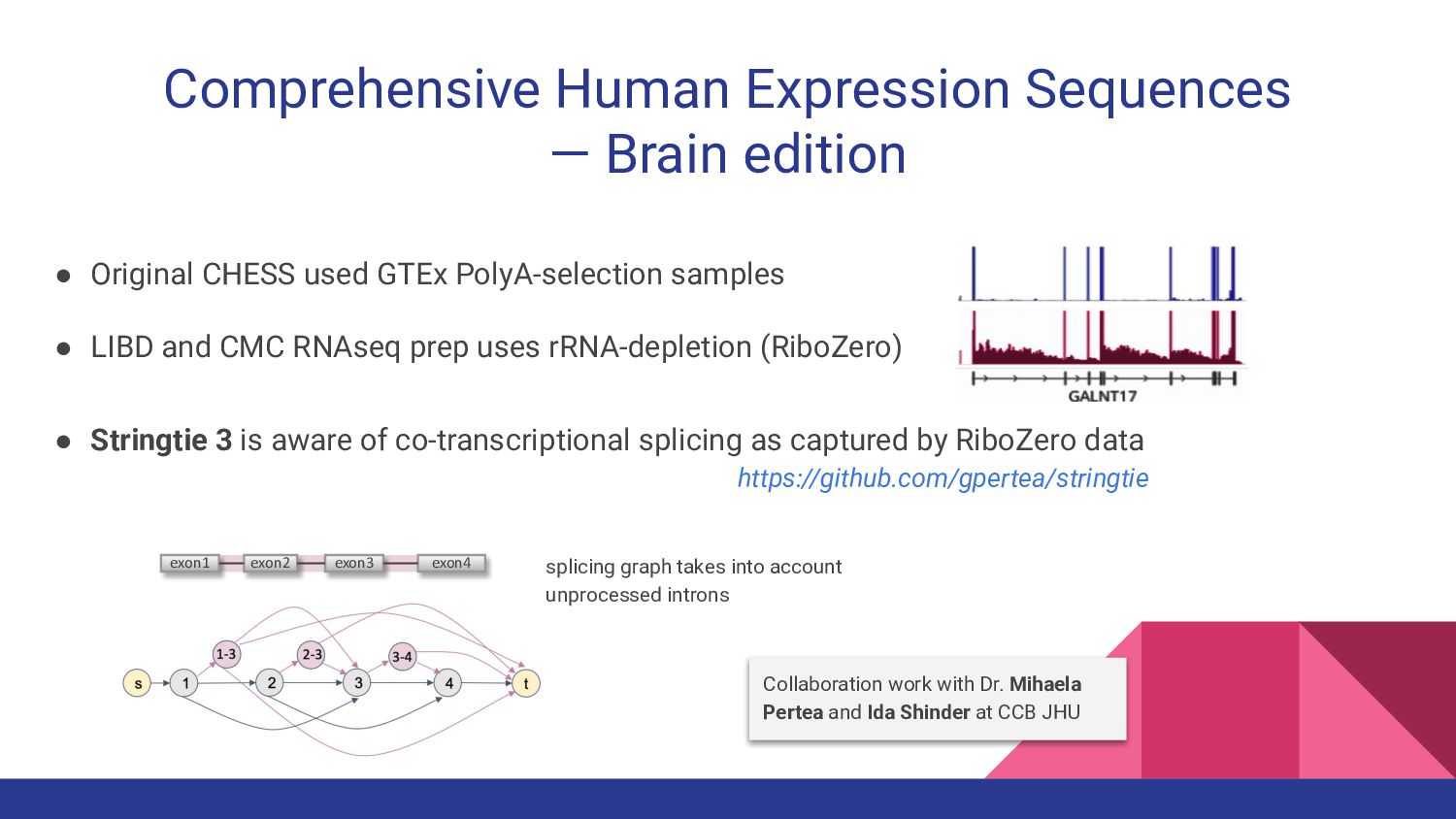
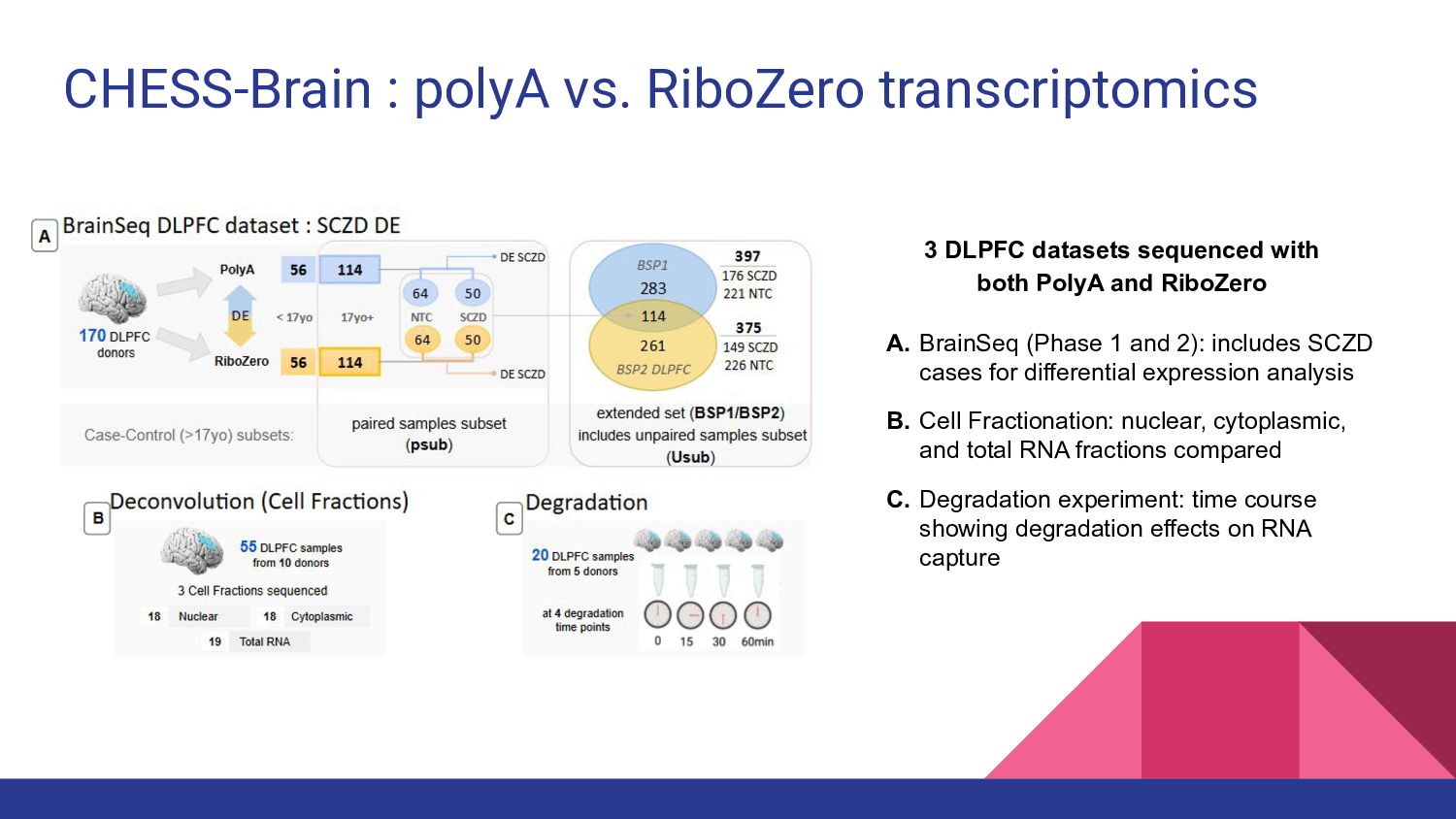
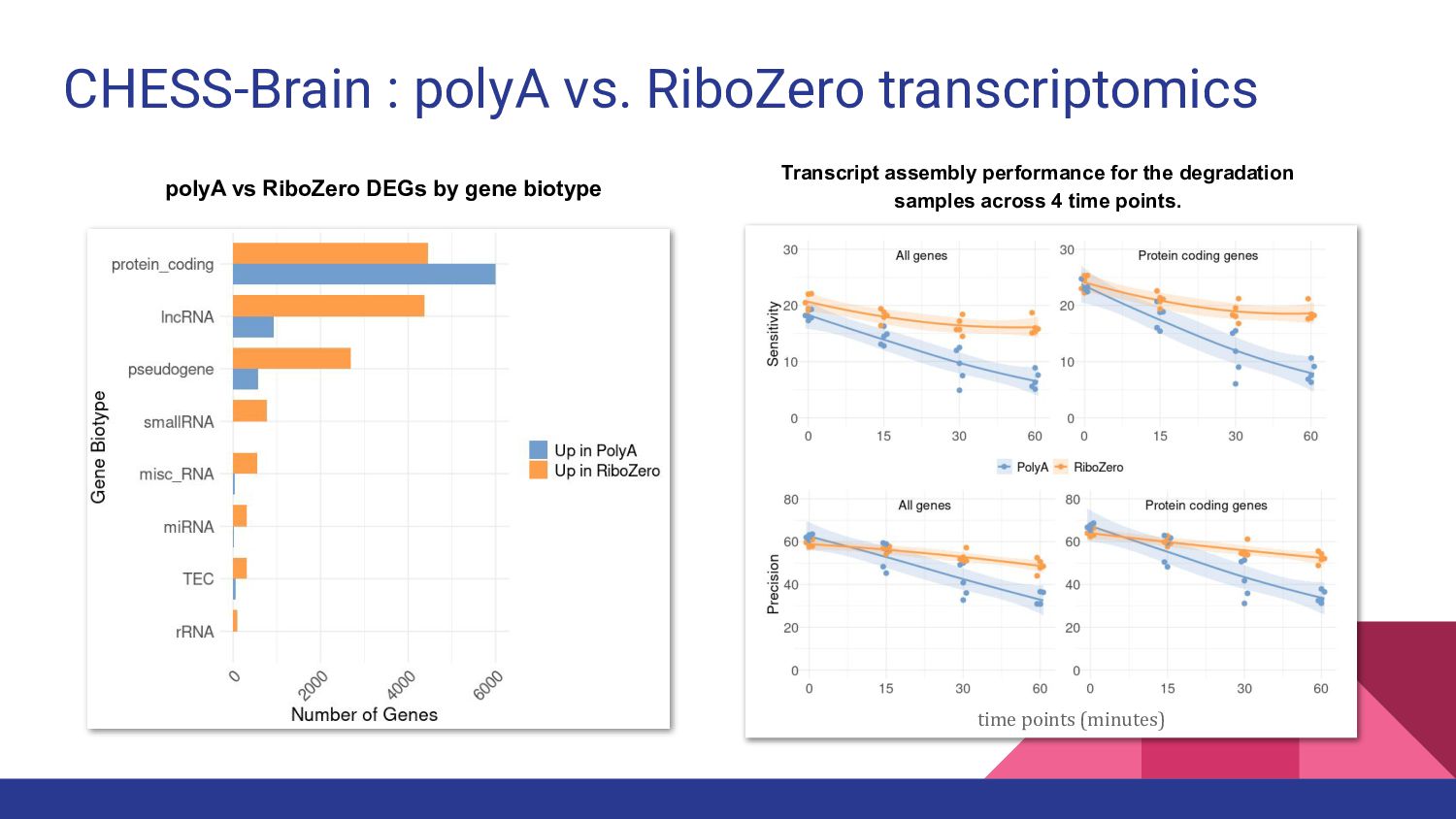
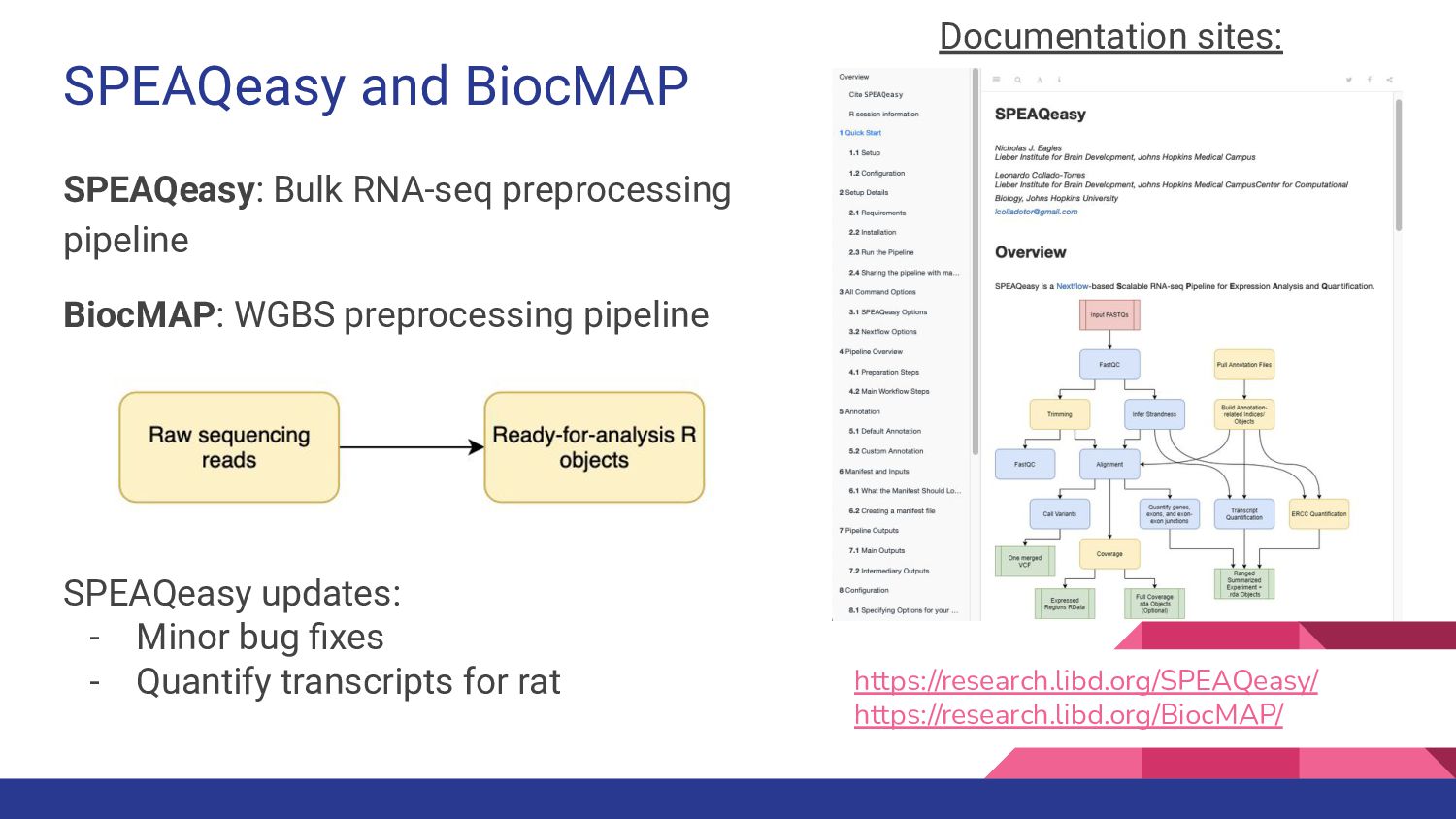
8. CHESS-Brain - Geo Pertea
9. Open Source Software Development - Nick Eagles
10. JHPCE - Nick Eagles
11. LIBD rstats & journal clubs - Nick Eages
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}