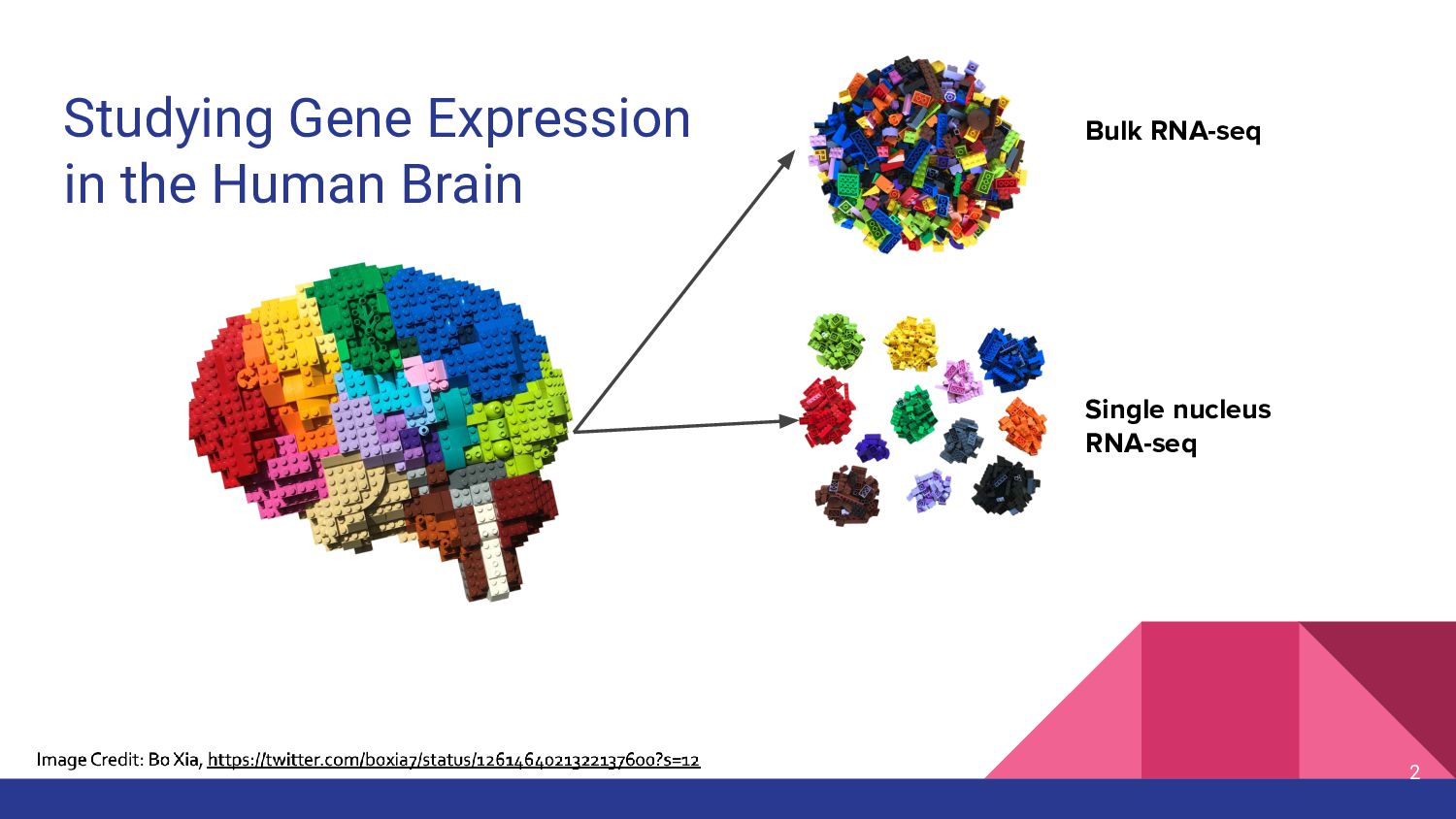
Benchmarking Deconvolution Methods in the Human Brain
Internal Seminar at the Lieber Institute for Brain Development (4/23/2024)
Presenting work from our Deconvolution Benchmark Preprint: https://doi.org/10.1101/2024.02.09.579665
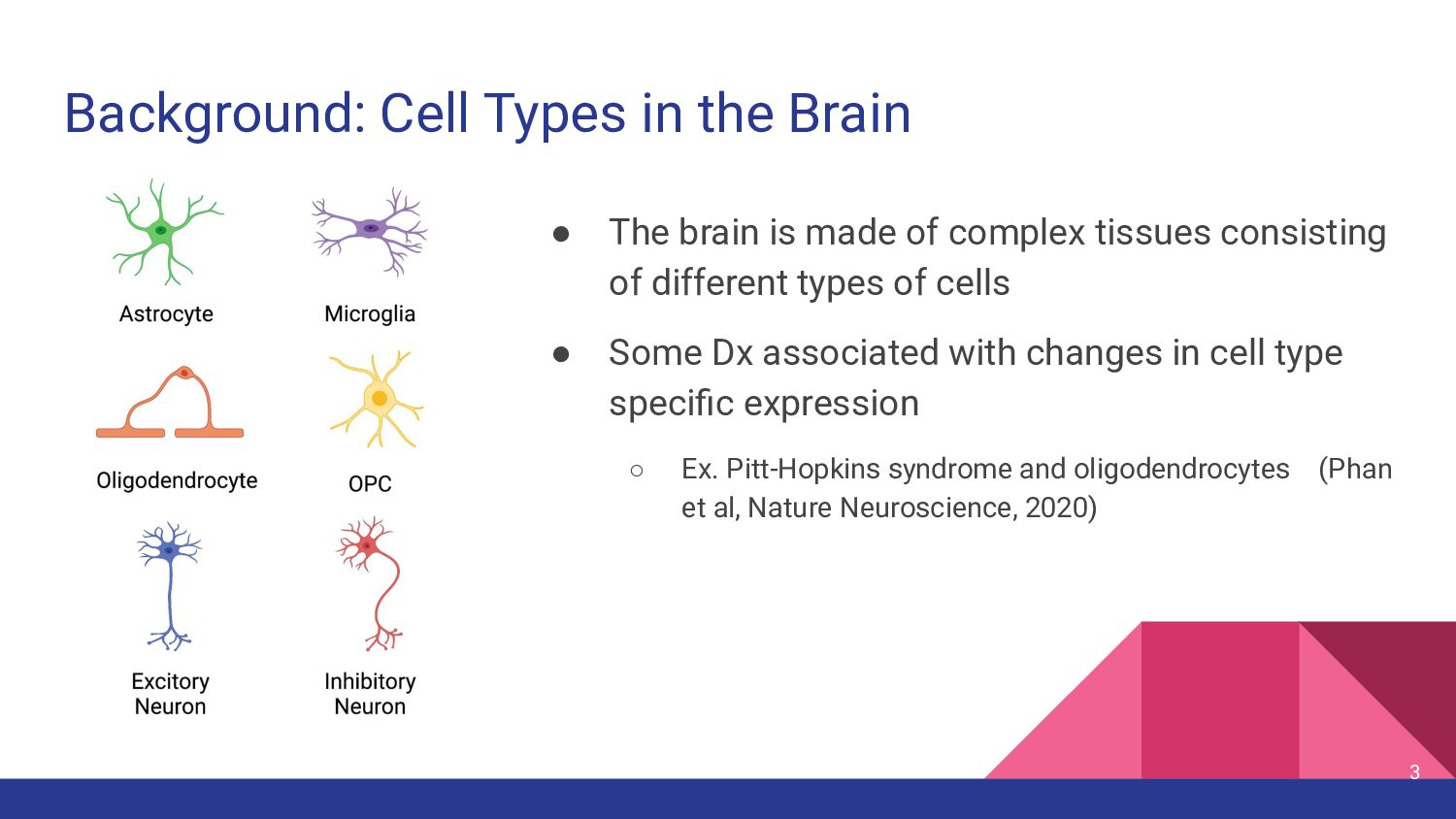
made of complex tissues consisting of different types of cells • Some Dx associated with changes in cell type specific expression ◦ Ex. Pitt-Hopkins syndrome and oligodendrocytes (Phan et al, Nature Neuroscience, 2020) 3
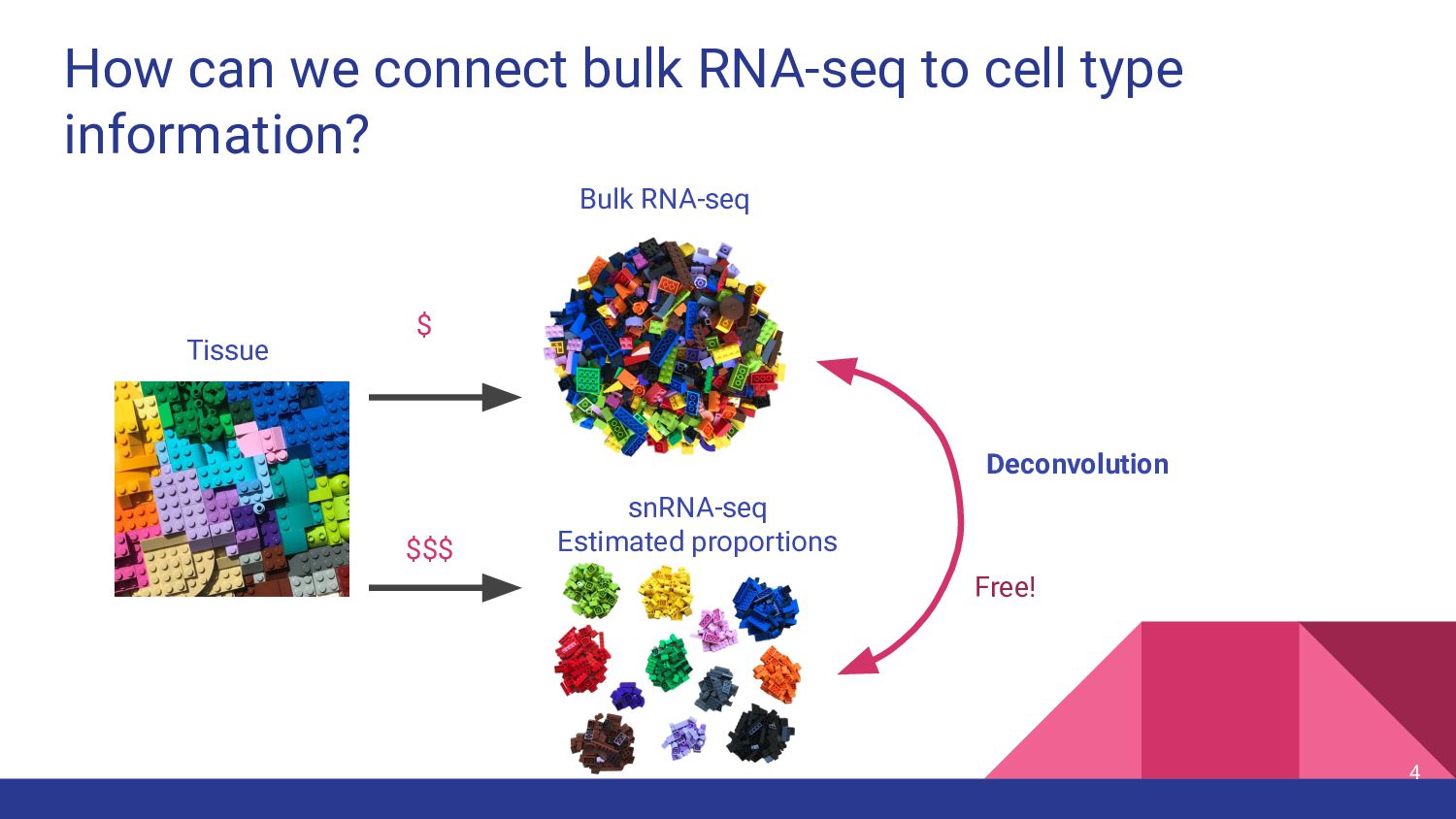
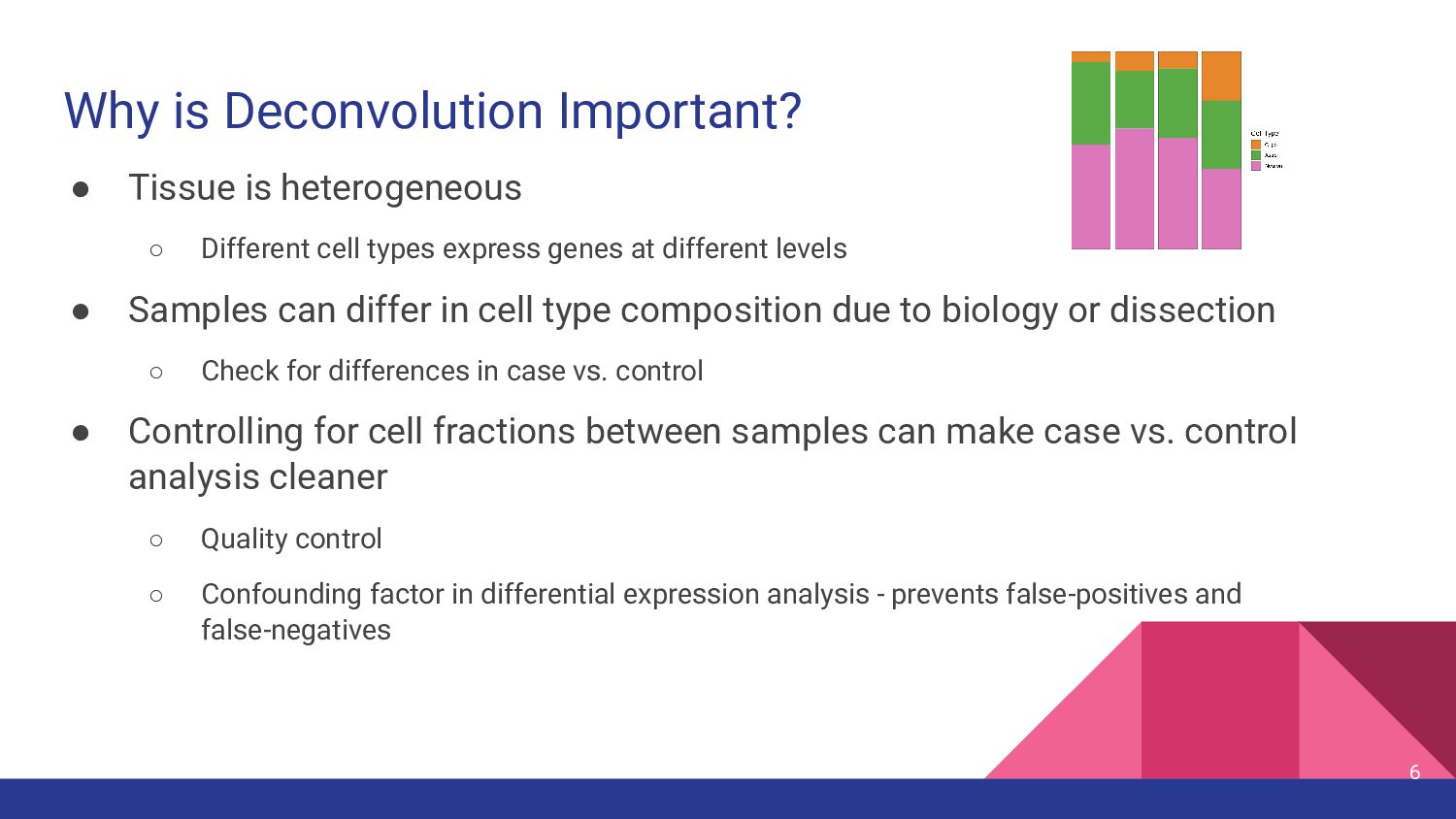
cell types express genes at different levels • Samples can differ in cell type composition due to biology or dissection ◦ Check for differences in case vs. control • Controlling for cell fractions between samples can make case vs. control analysis cleaner ◦ Quality control ◦ Confounding factor in differential expression analysis - prevents false-positives and false-negatives 6
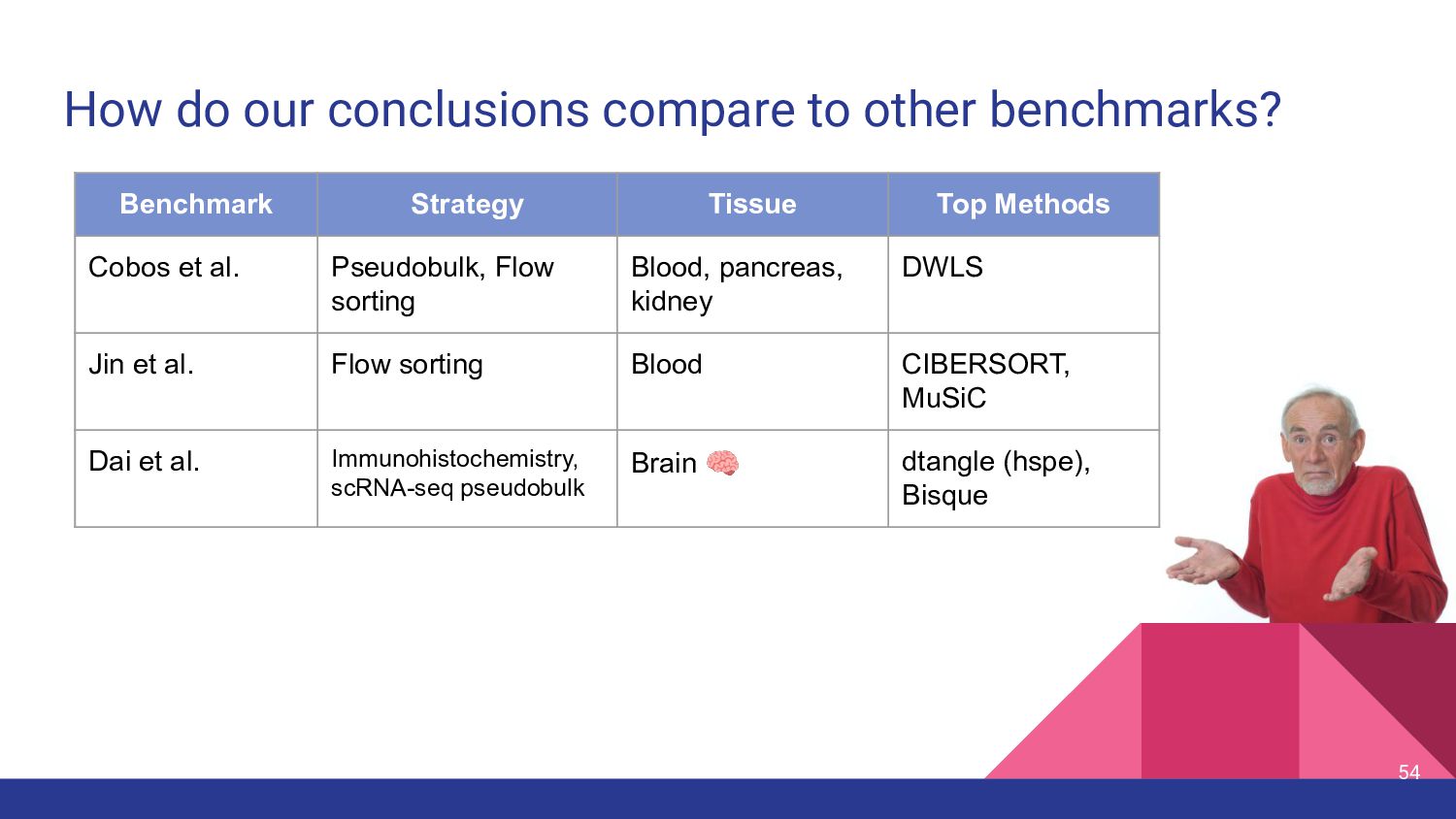
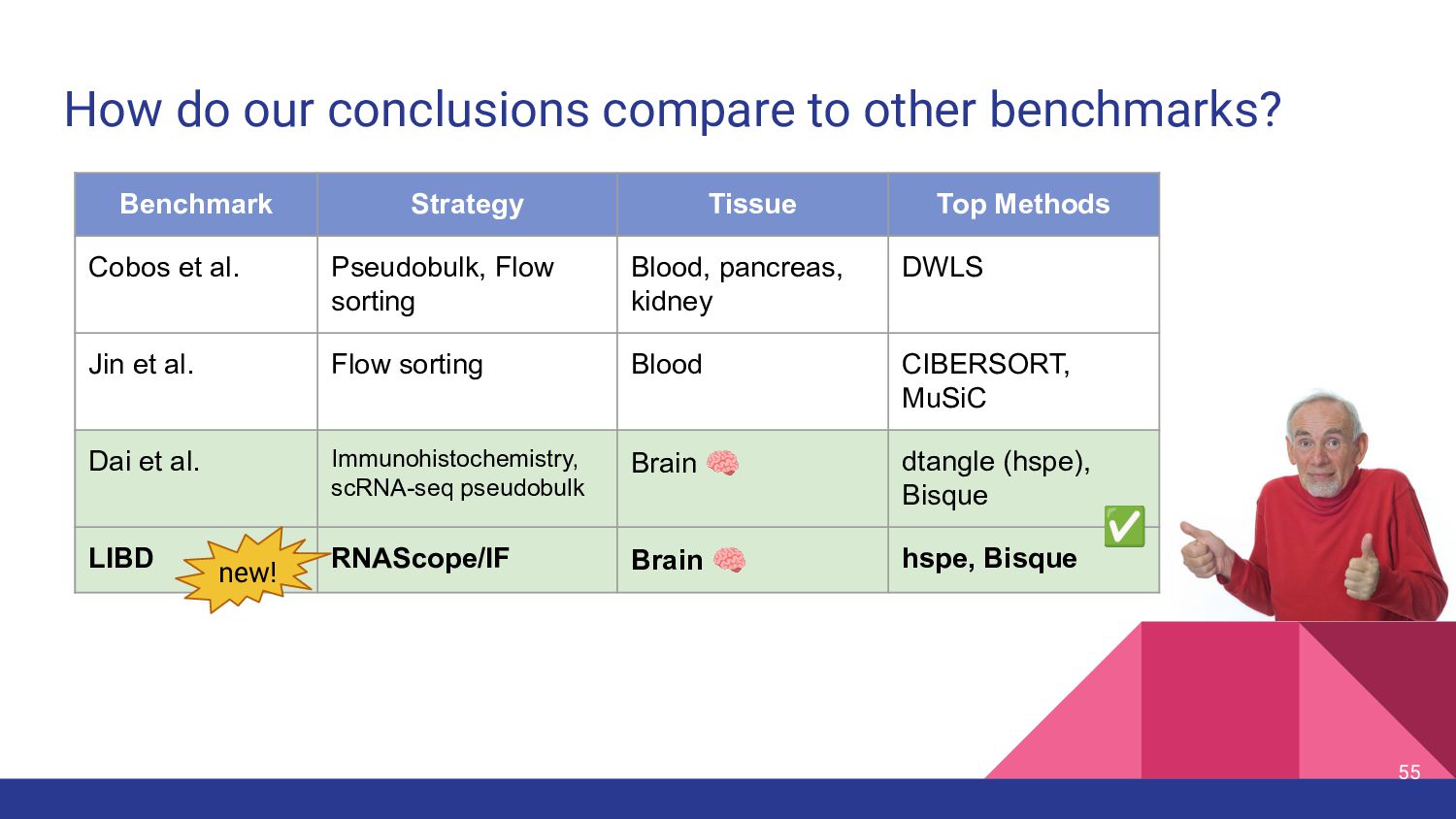
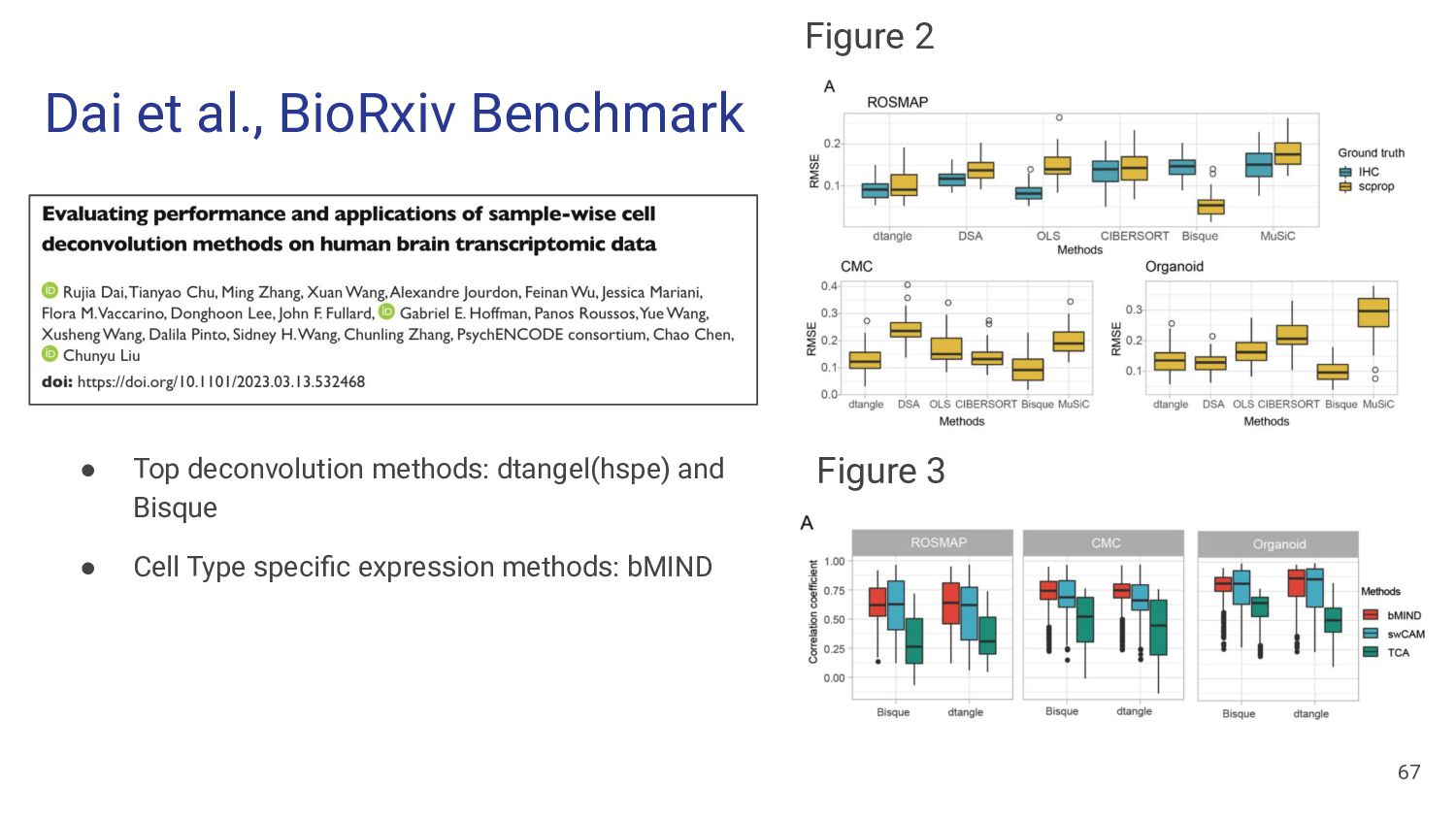
different methods perform best on different data sets (Cobos et al, Nature Communications, 2020) • Benchmarking results from different papers on “real” data ◦ MuSiC paper: MuSiC > NNLS > BSEQ-sx > CIBERSORT ▪ Pancreatic Islet: Beta cells vs. HbA1c (Fig 2a) ◦ Bisque paper: Bisque > MuSiC > CIBERSORT ▪ DLPFC: Microglia vs. Braak stage, Neuron vs. Cognitive diagnostic category (Fig 4) ◦ Cobos benchmark: DWLS > MuSiC > Bisque > deconvoSeq ▪ Human PMBC flow sorted (Fig 7) ◦ Jin et al. benchmark: CIBERSORT, MuSiC > EPIC*, TIMER, DeconRNAseq ▪ Human Whole Blood, simulations ◦ Dai et al., benchmark: Dtangle > Bisque > Other Methods ▪ human brain IHC & scRNA-seq data 9
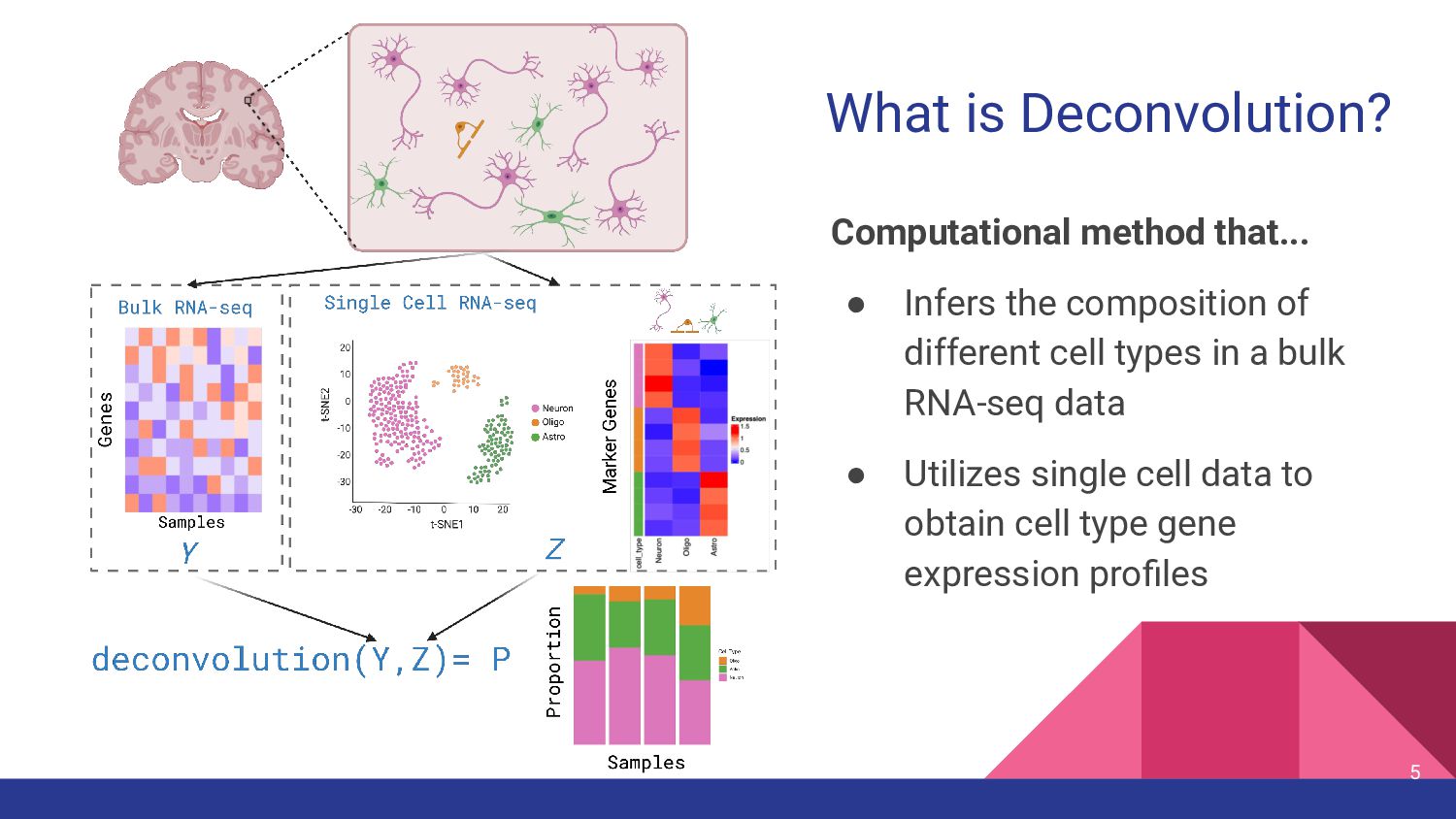
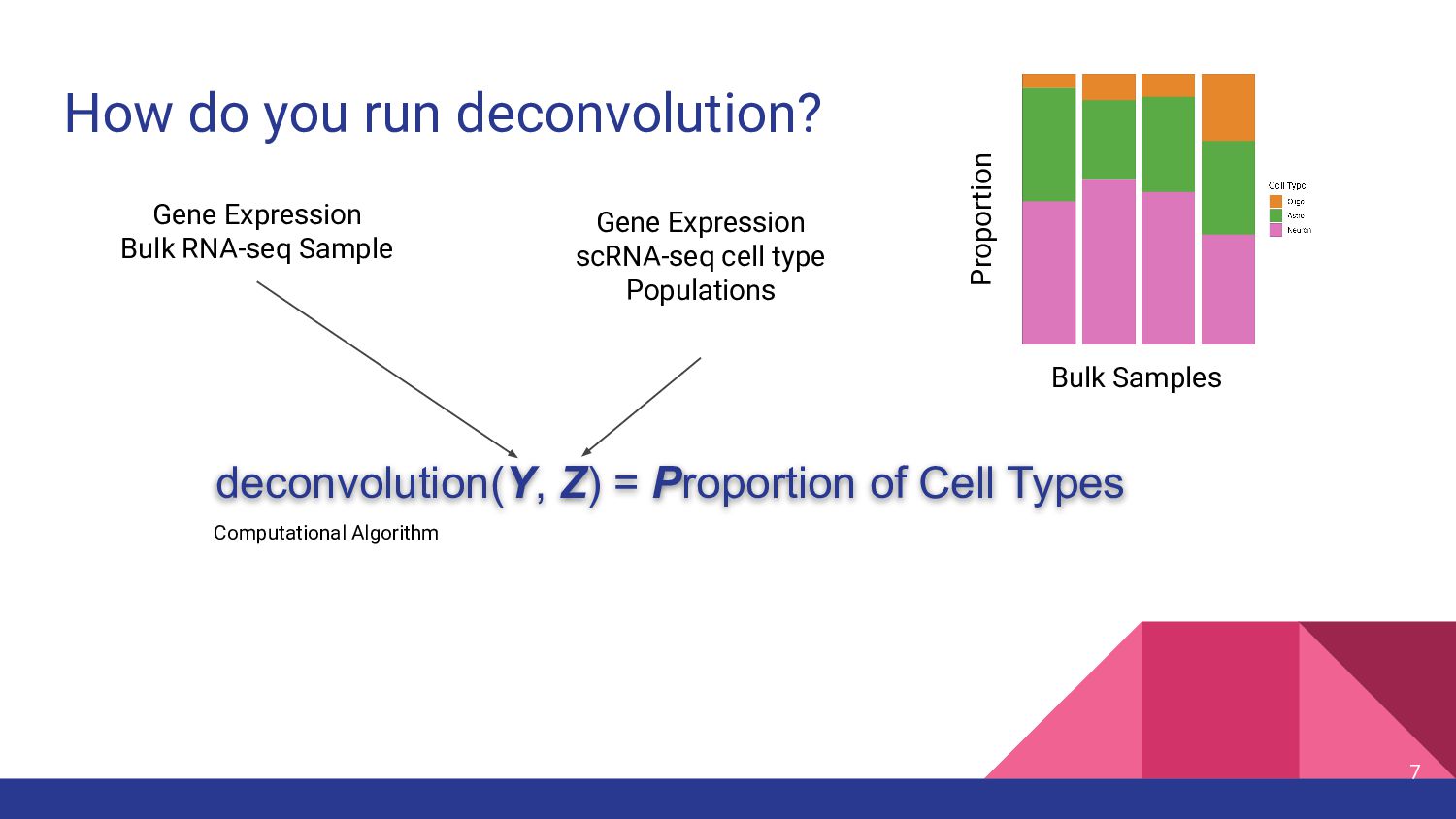
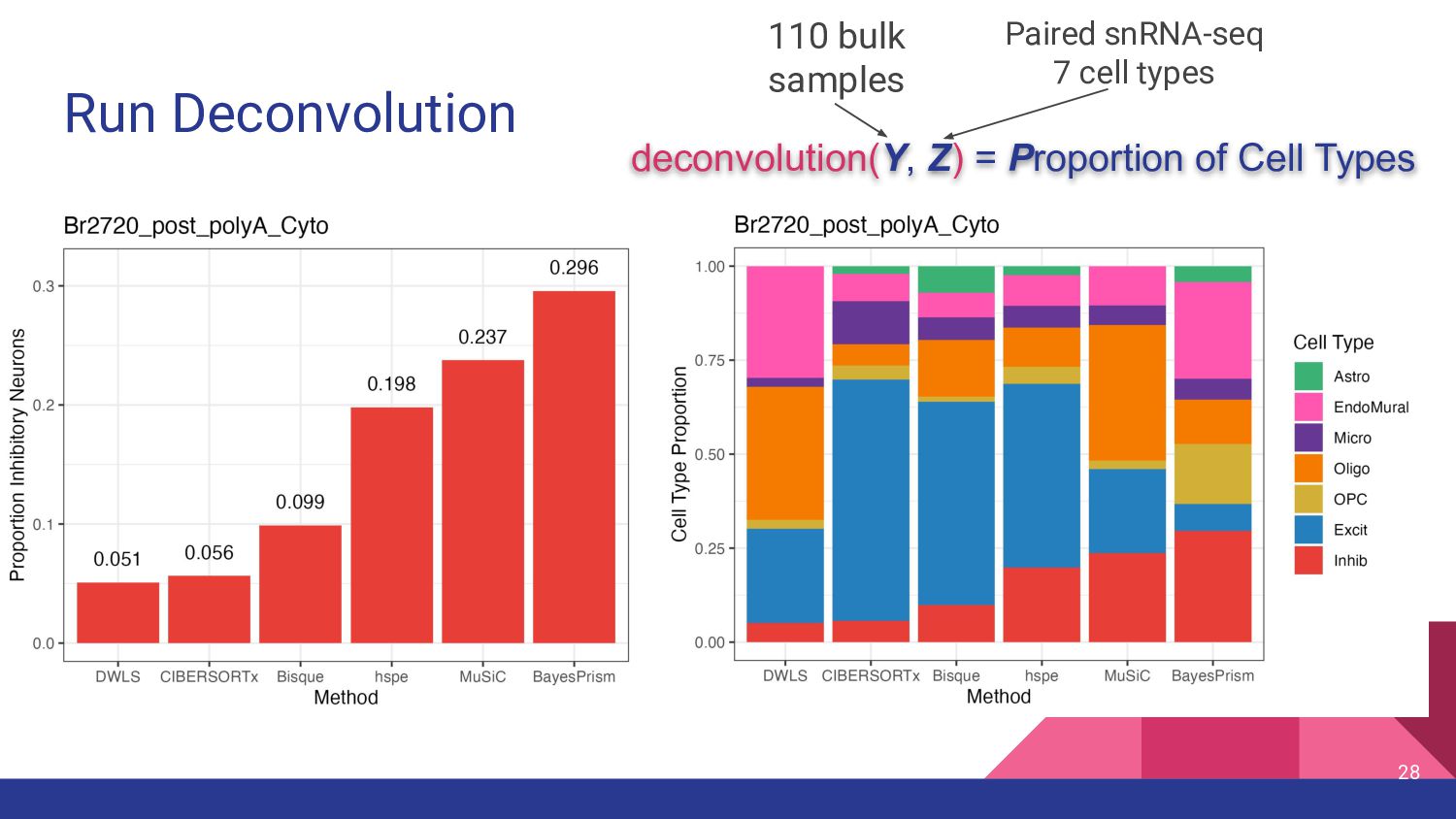
cell type measurements • Test top deconvolution methods that employ different strategies • Assess impact of other factors in deconvolution ◦ Bulk RNA-seq data types ◦ snRNA-seq features ◦ Marker genes 11
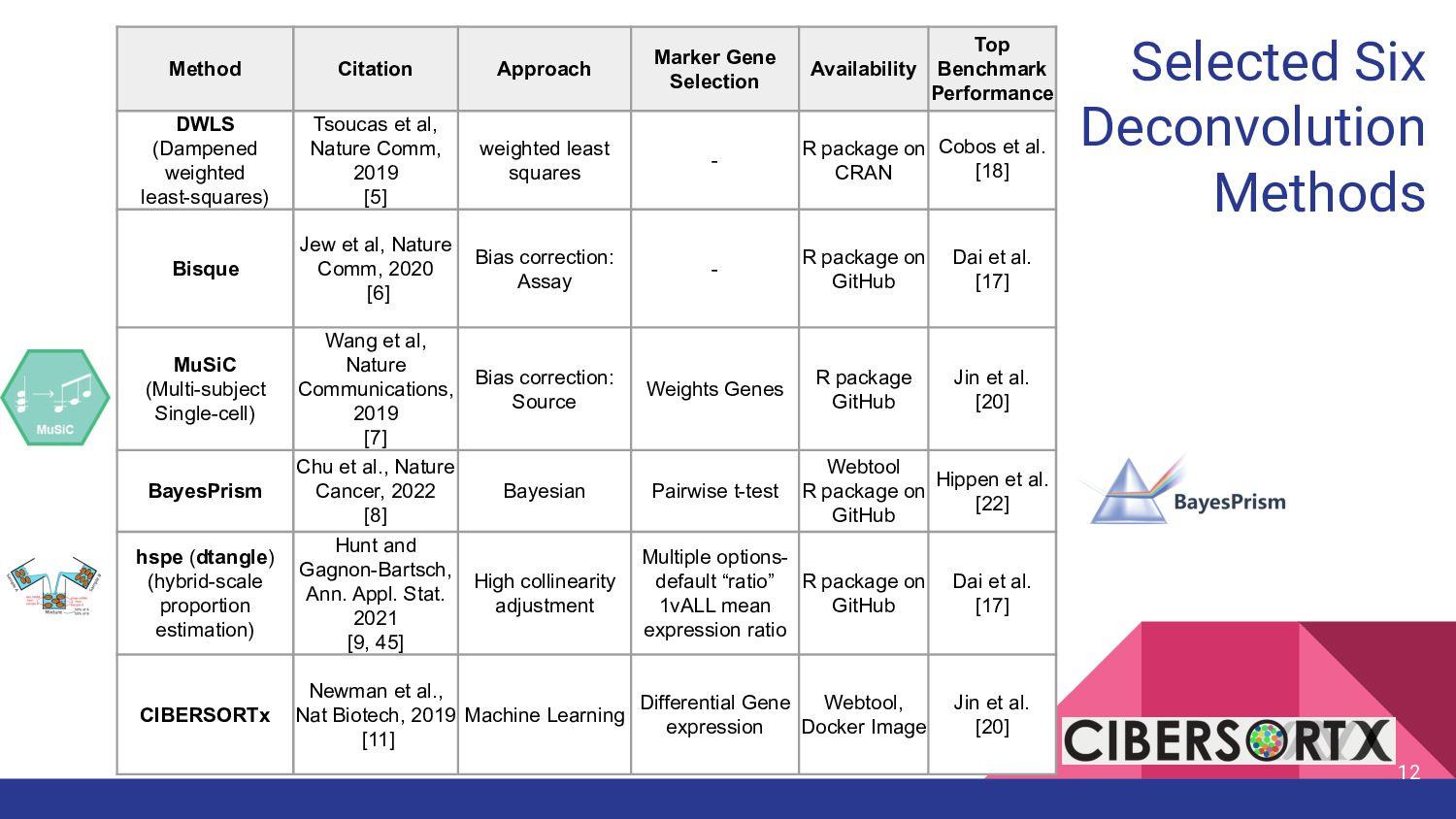
Selection Availability Top Benchmark Performance DWLS (Dampened weighted least-squares) Tsoucas et al, Nature Comm, 2019 [5] weighted least squares - R package on CRAN Cobos et al. [18] Bisque Jew et al, Nature Comm, 2020 [6] Bias correction: Assay - R package on GitHub Dai et al. [17] MuSiC (Multi-subject Single-cell) Wang et al, Nature Communications, 2019 [7] Bias correction: Source Weights Genes R package GitHub Jin et al. [20] BayesPrism Chu et al., Nature Cancer, 2022 [8] Bayesian Pairwise t-test Webtool R package on GitHub Hippen et al. [22] hspe (dtangle) (hybrid-scale proportion estimation) Hunt and Gagnon-Bartsch, Ann. Appl. Stat. 2021 [9, 45] High collinearity adjustment Multiple options- default “ratio” 1vALL mean expression ratio R package on GitHub Dai et al. [17] CIBERSORTx Newman et al., Nat Biotech, 2019 [11] Machine Learning Differential Gene expression Webtool, Docker Image Jin et al. [20]
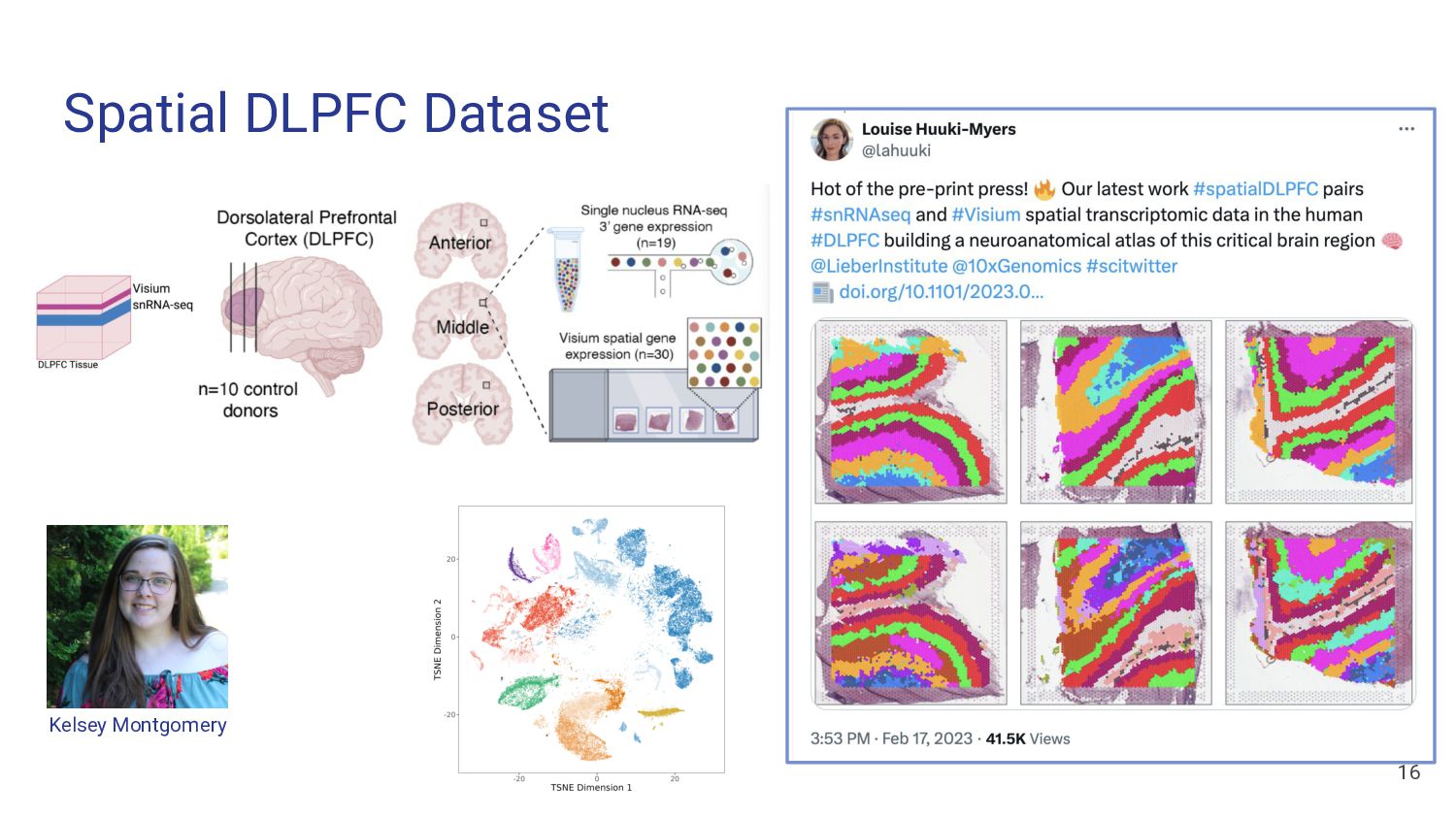
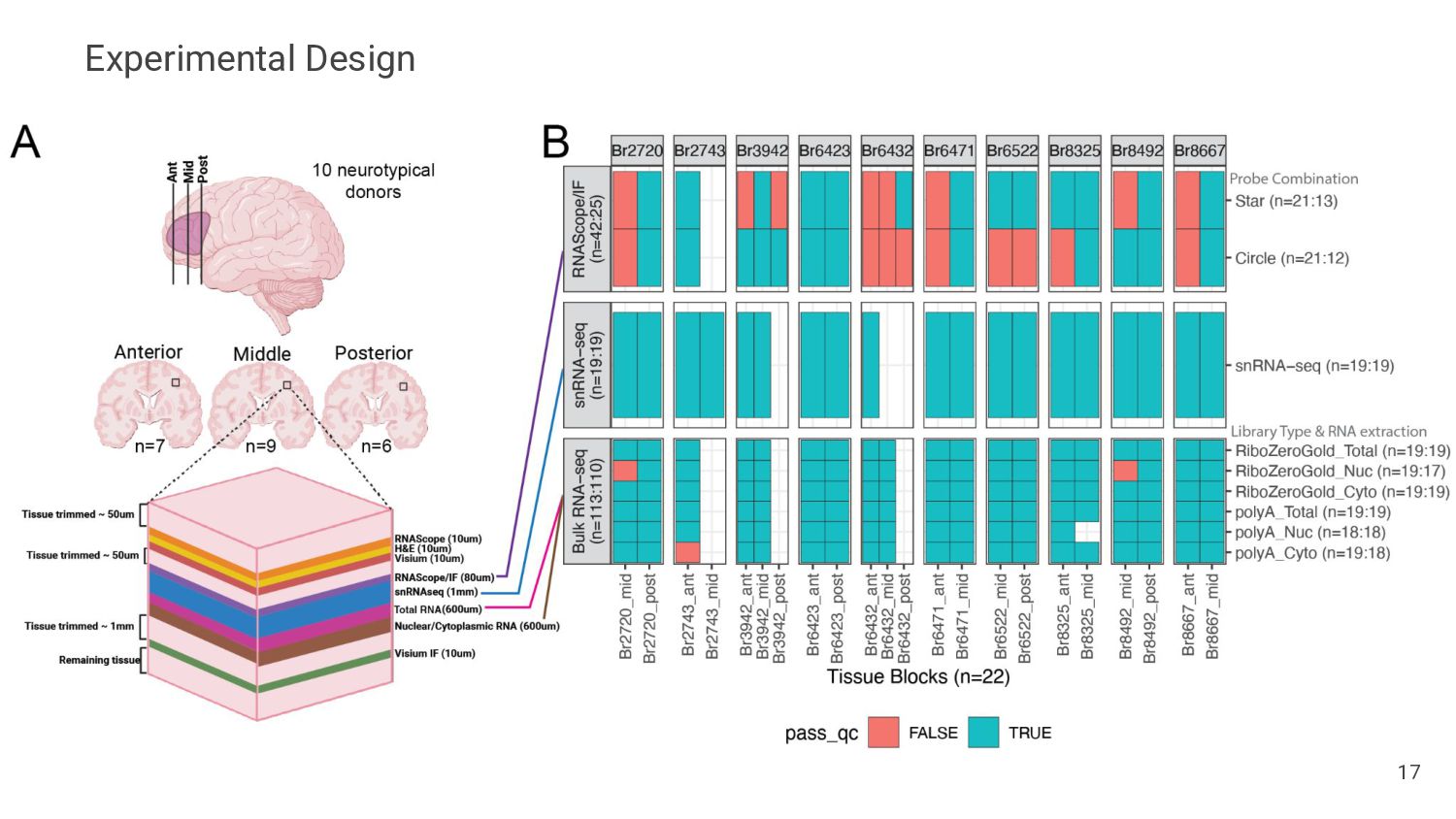
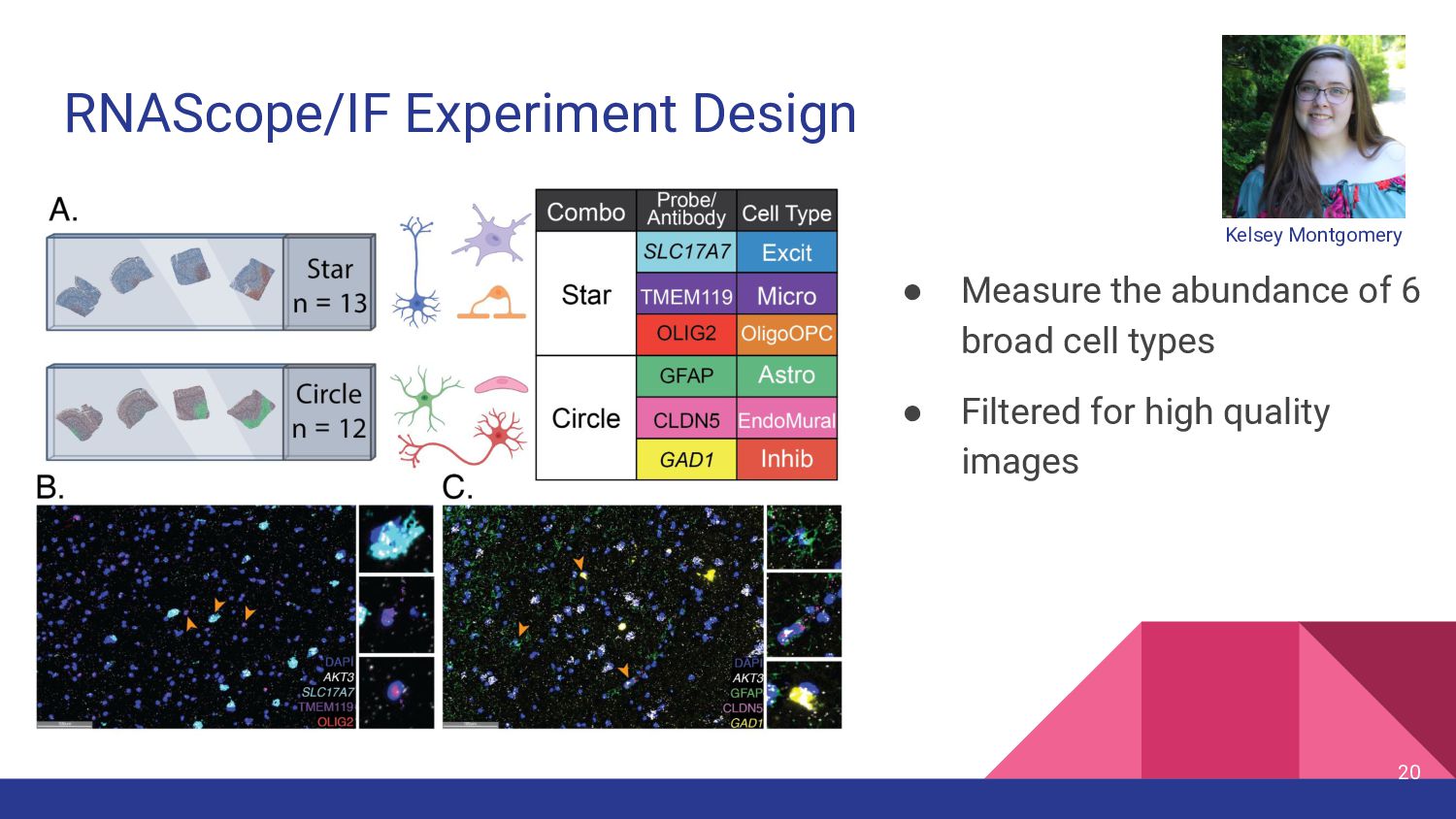
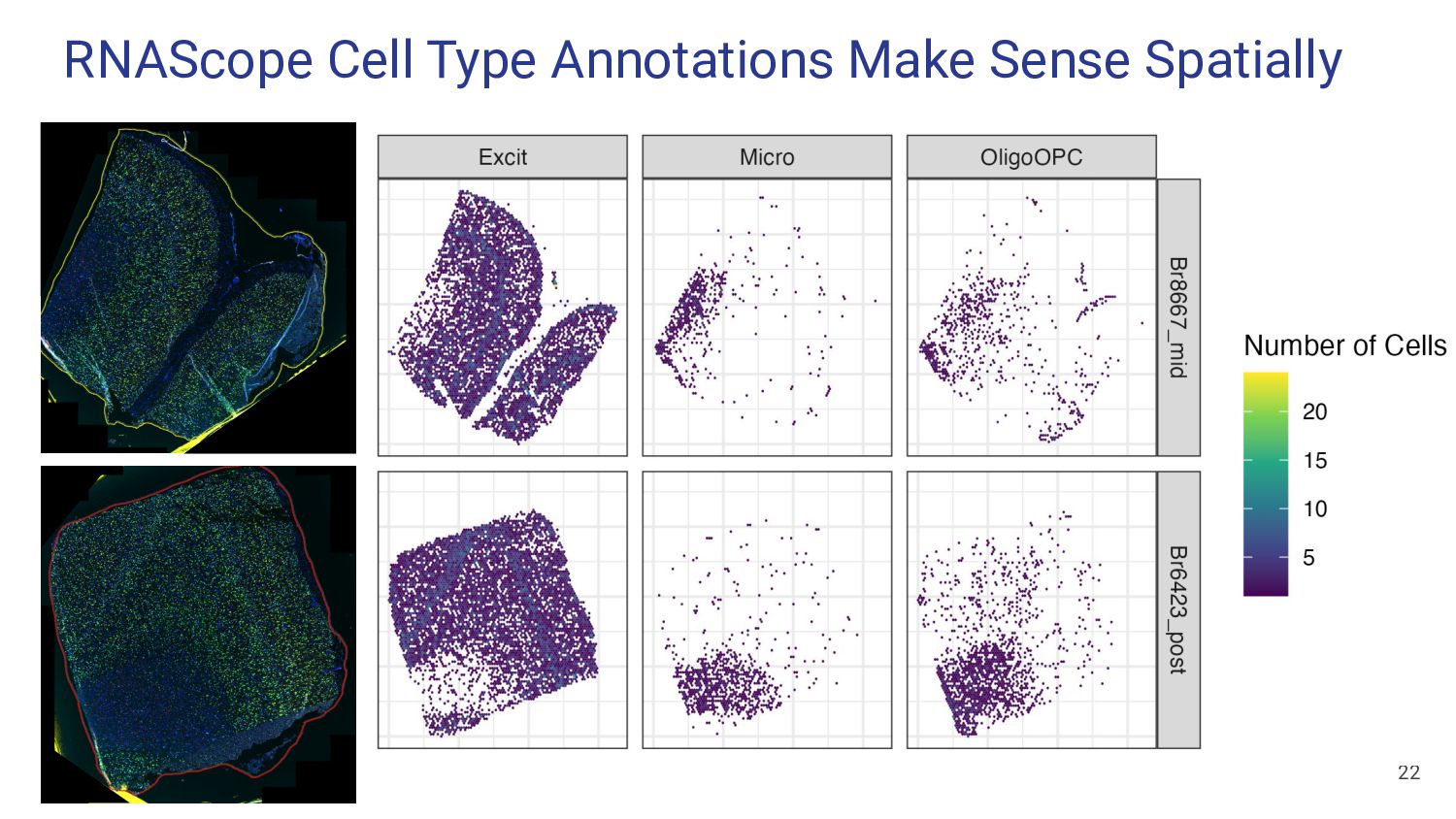
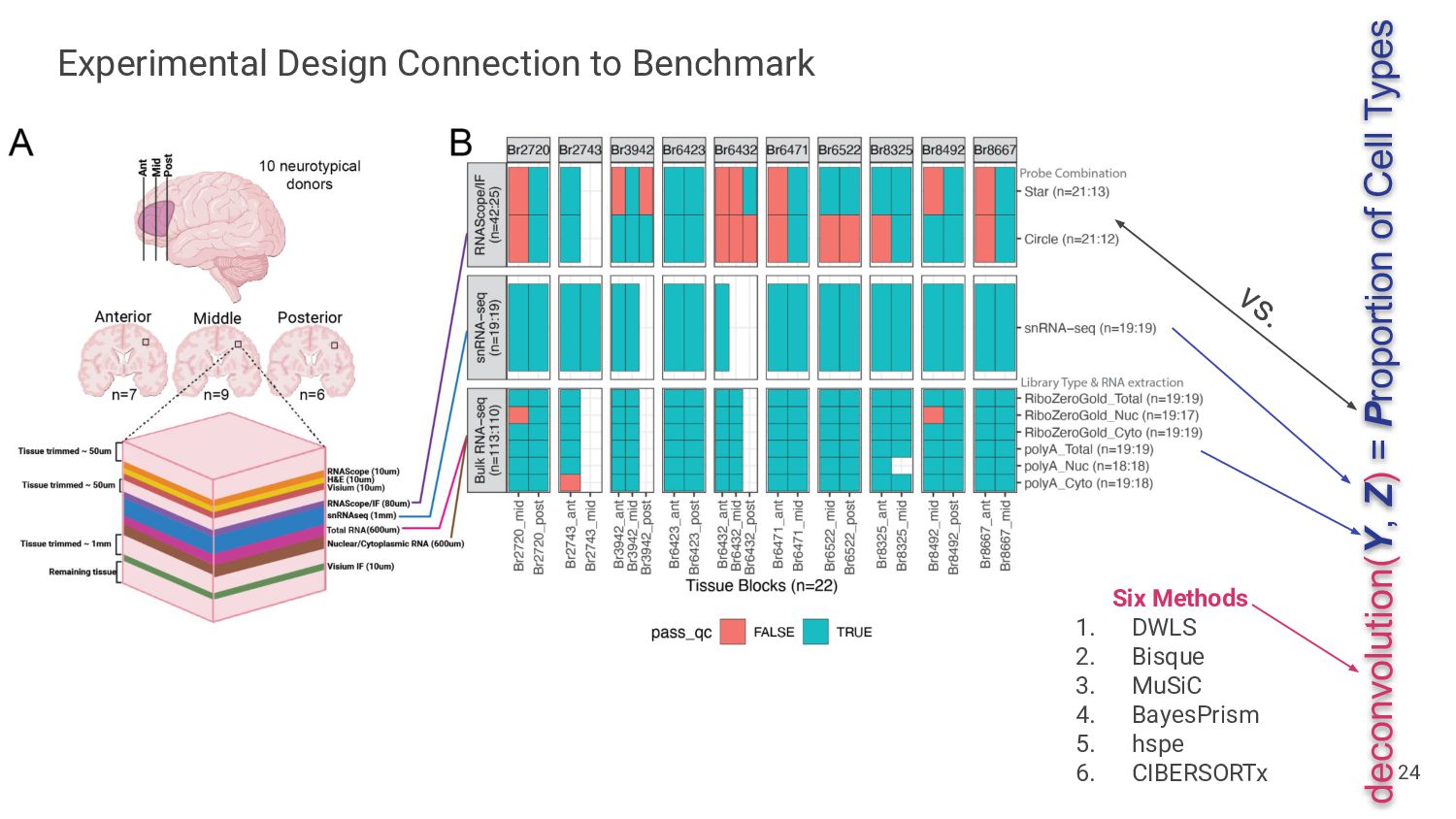
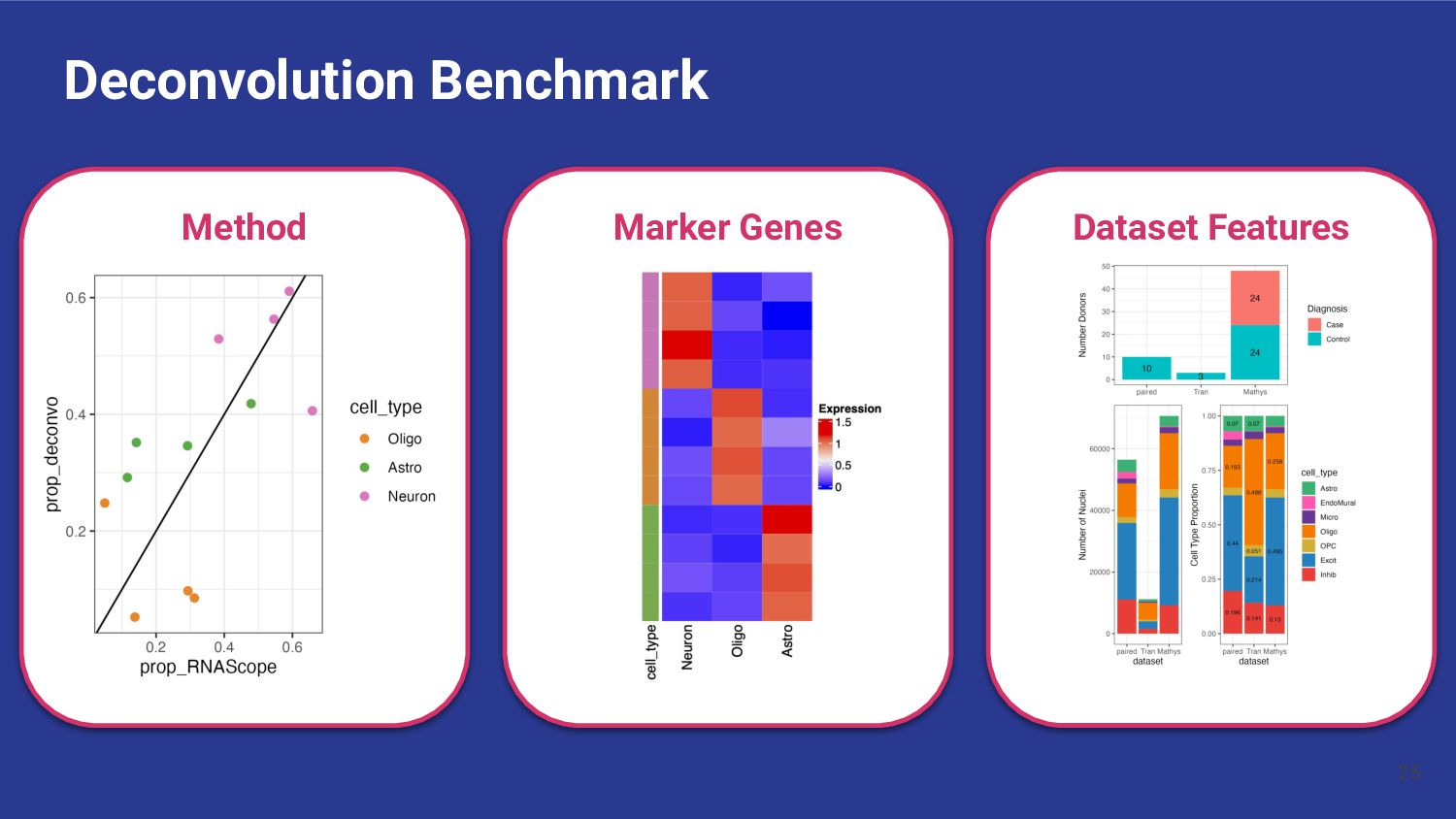
Assess Accuracy • Use pseudobulk samples ◦ Known or simulated composition ◦ May not reflect real bulk RNA-seq data • Compare with Immunofluorescence Data • Cell flow sorting ◦ Difficult to label nuclei by cell type 13 Our Strategy • Use paired orthogonal imaging data to measure cell type proportions & evaluate method accuracy • Focus on brain tissue
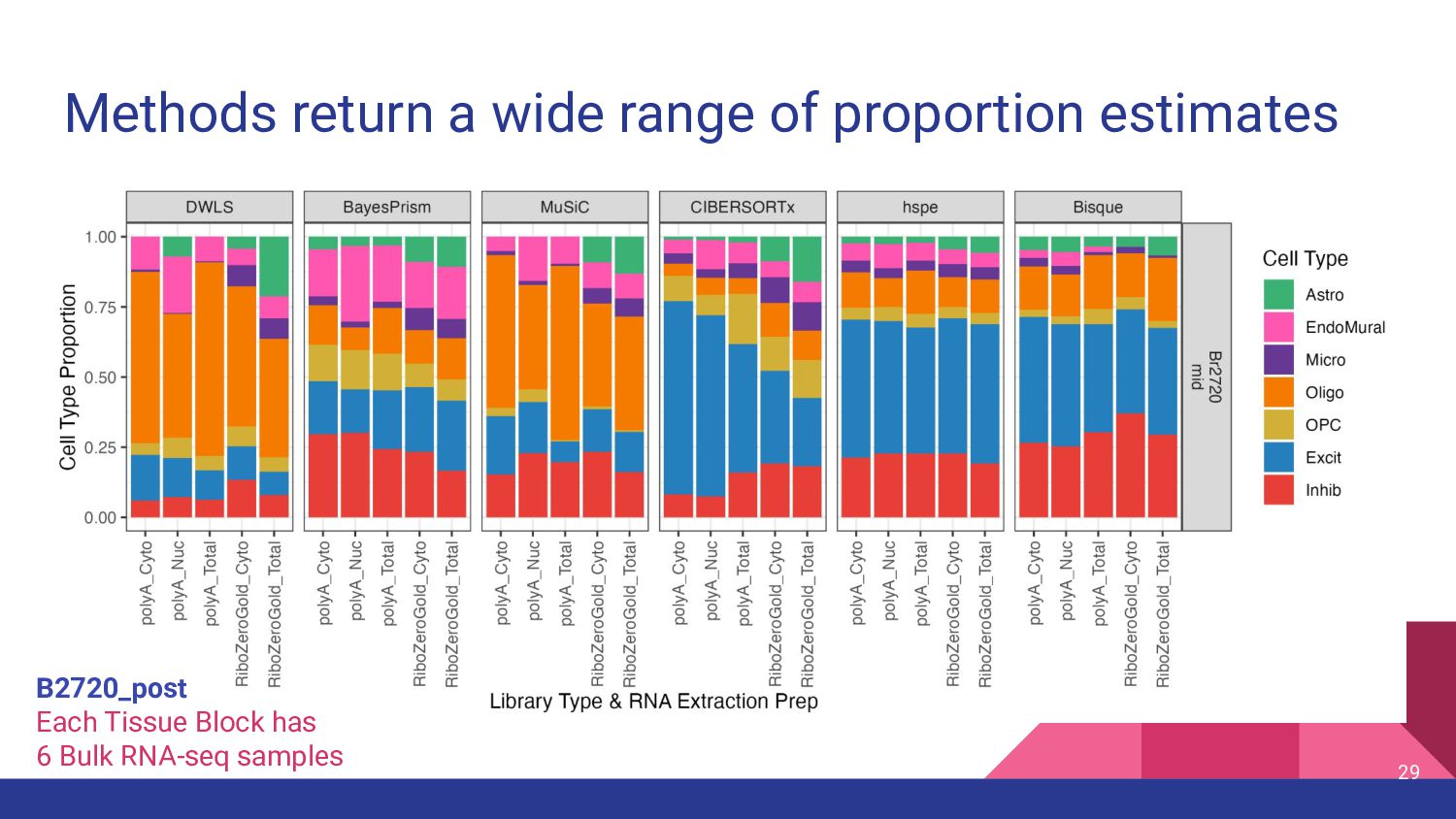
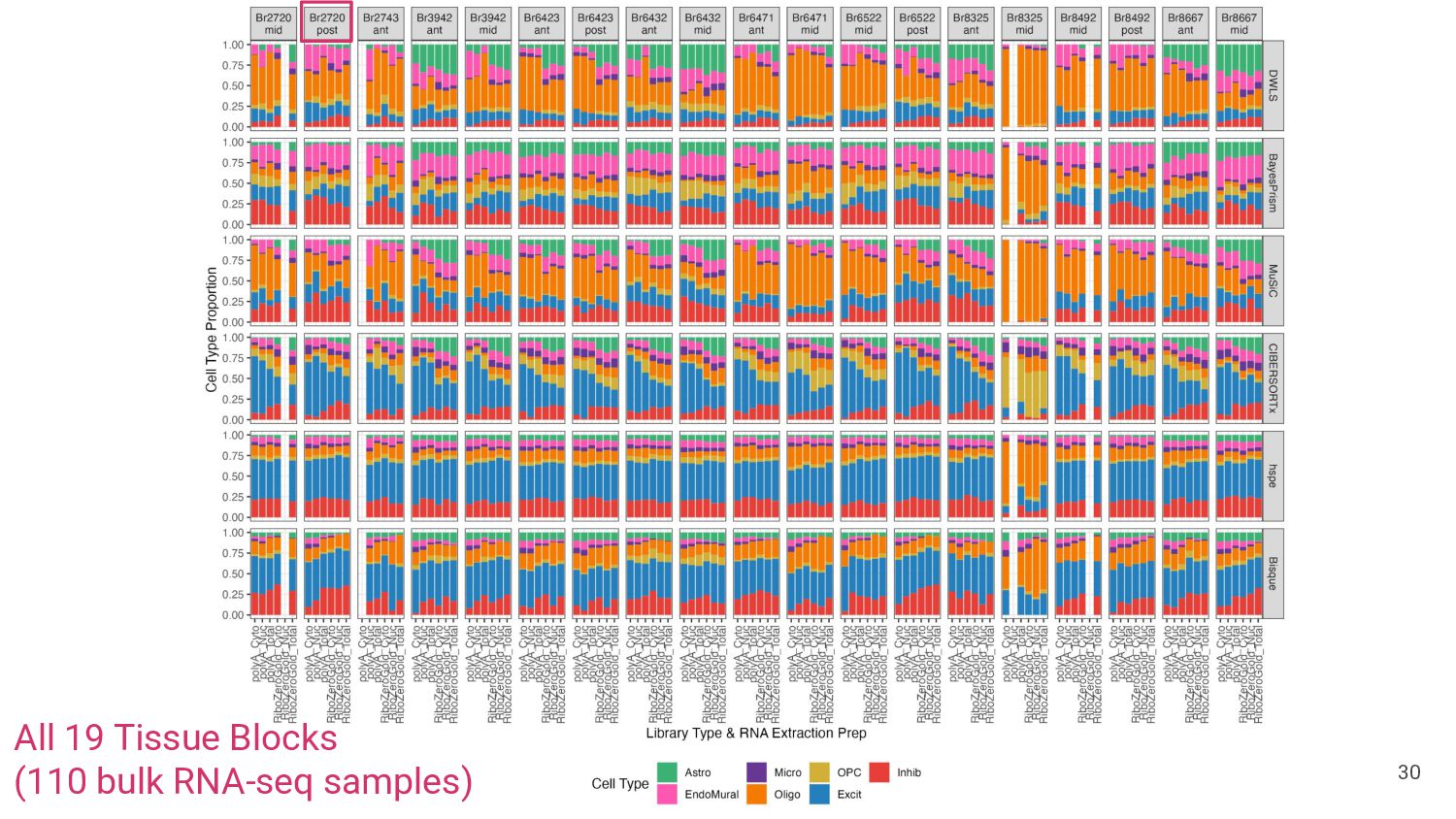
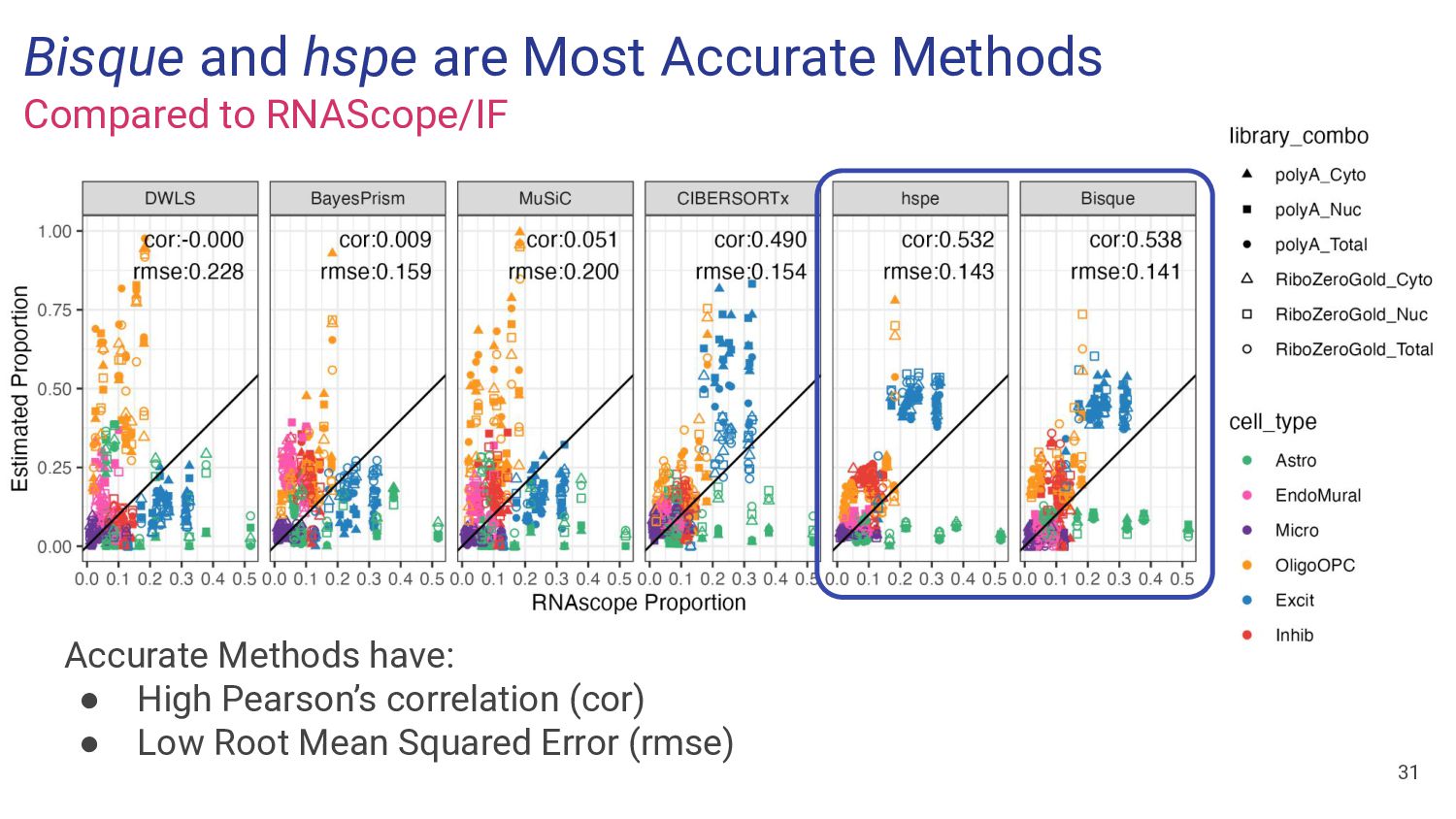
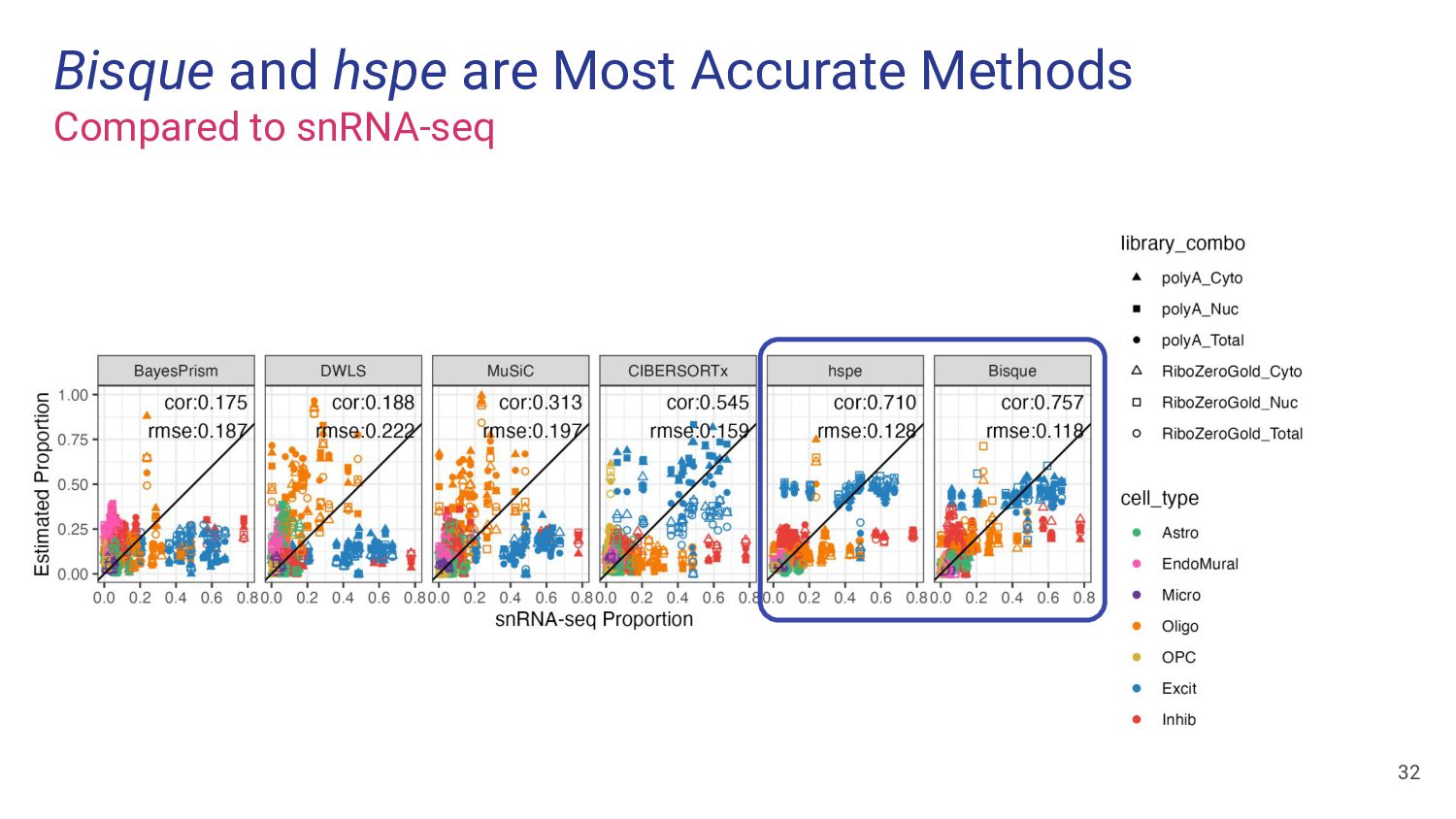
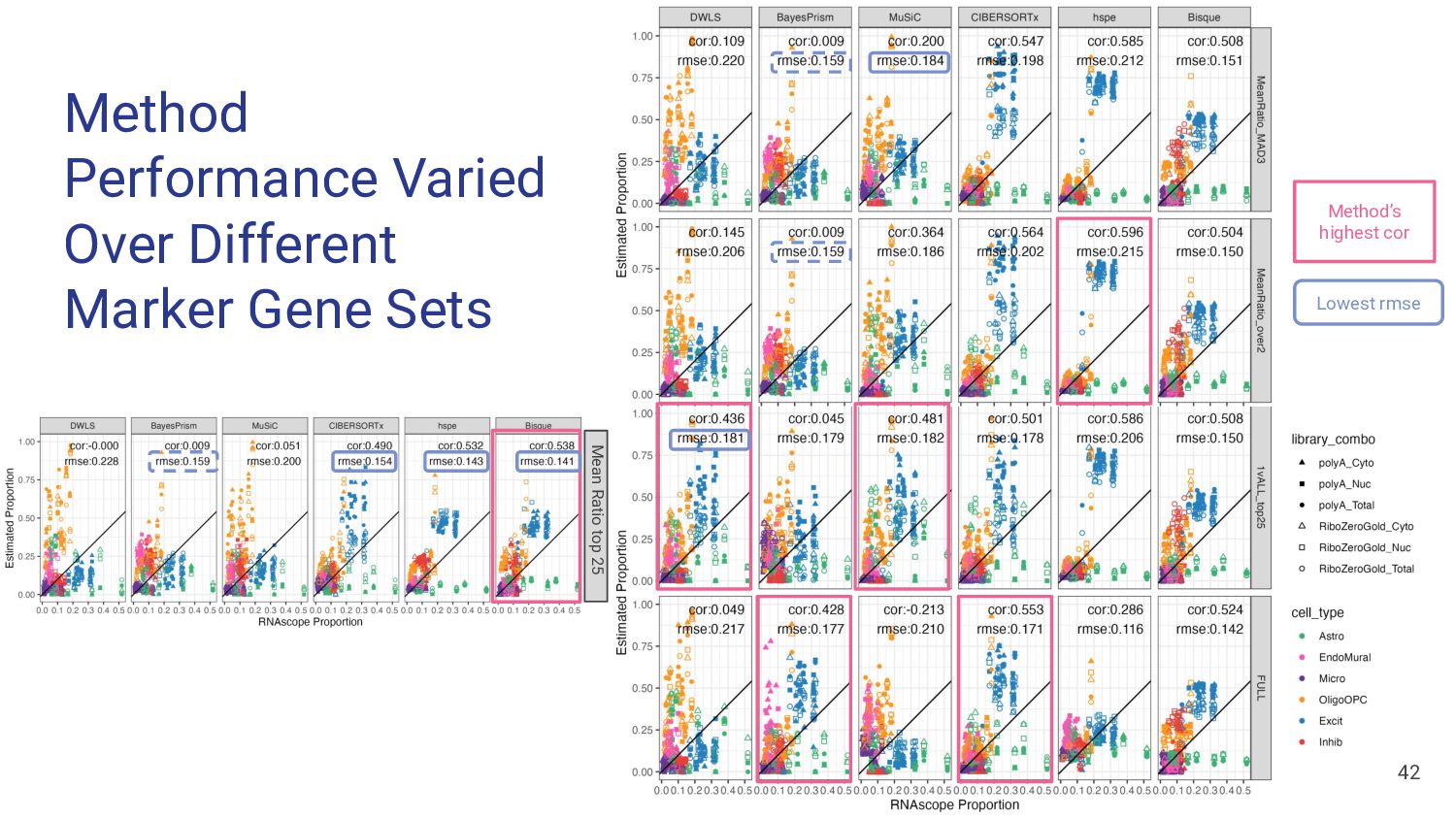
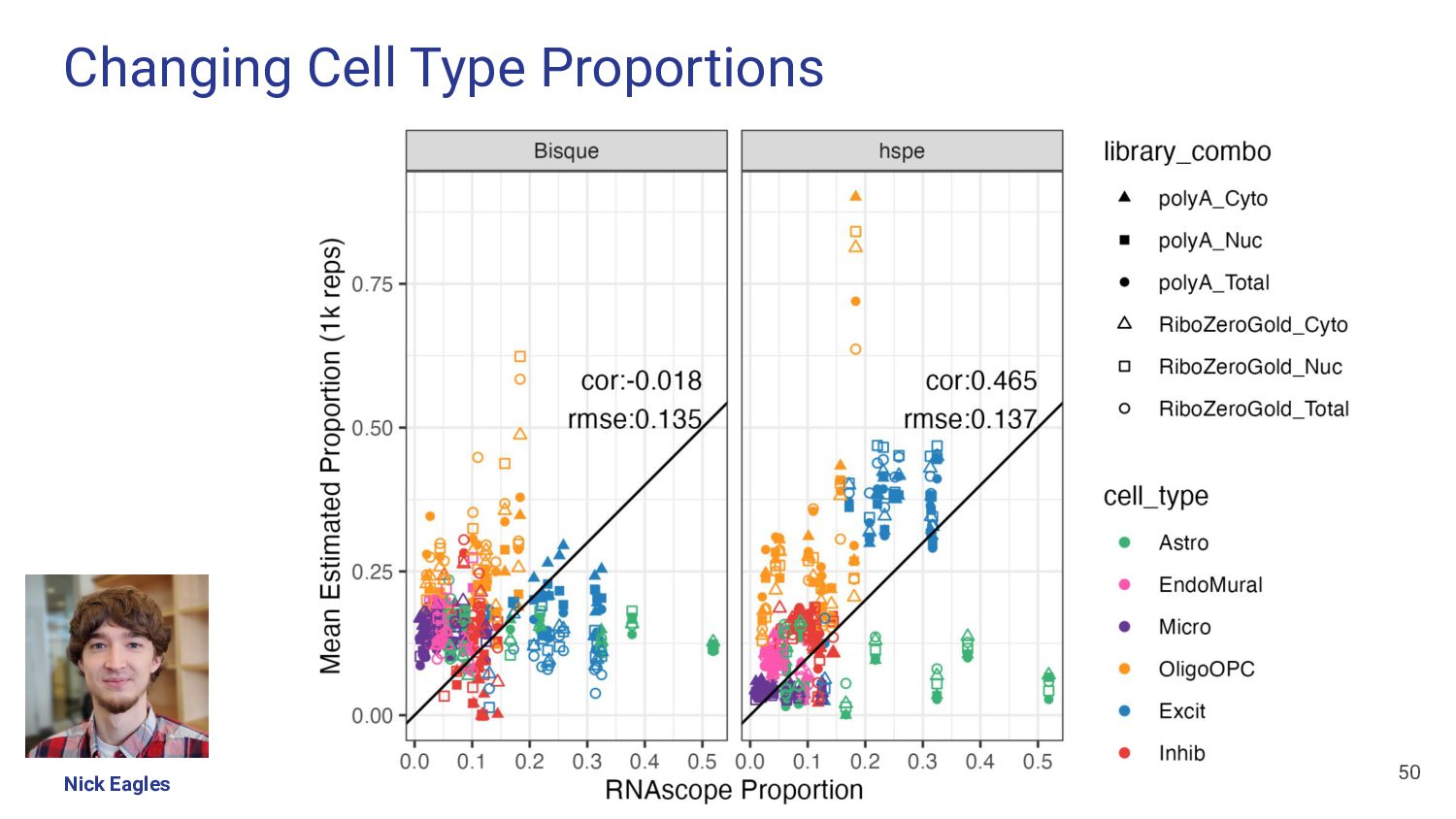
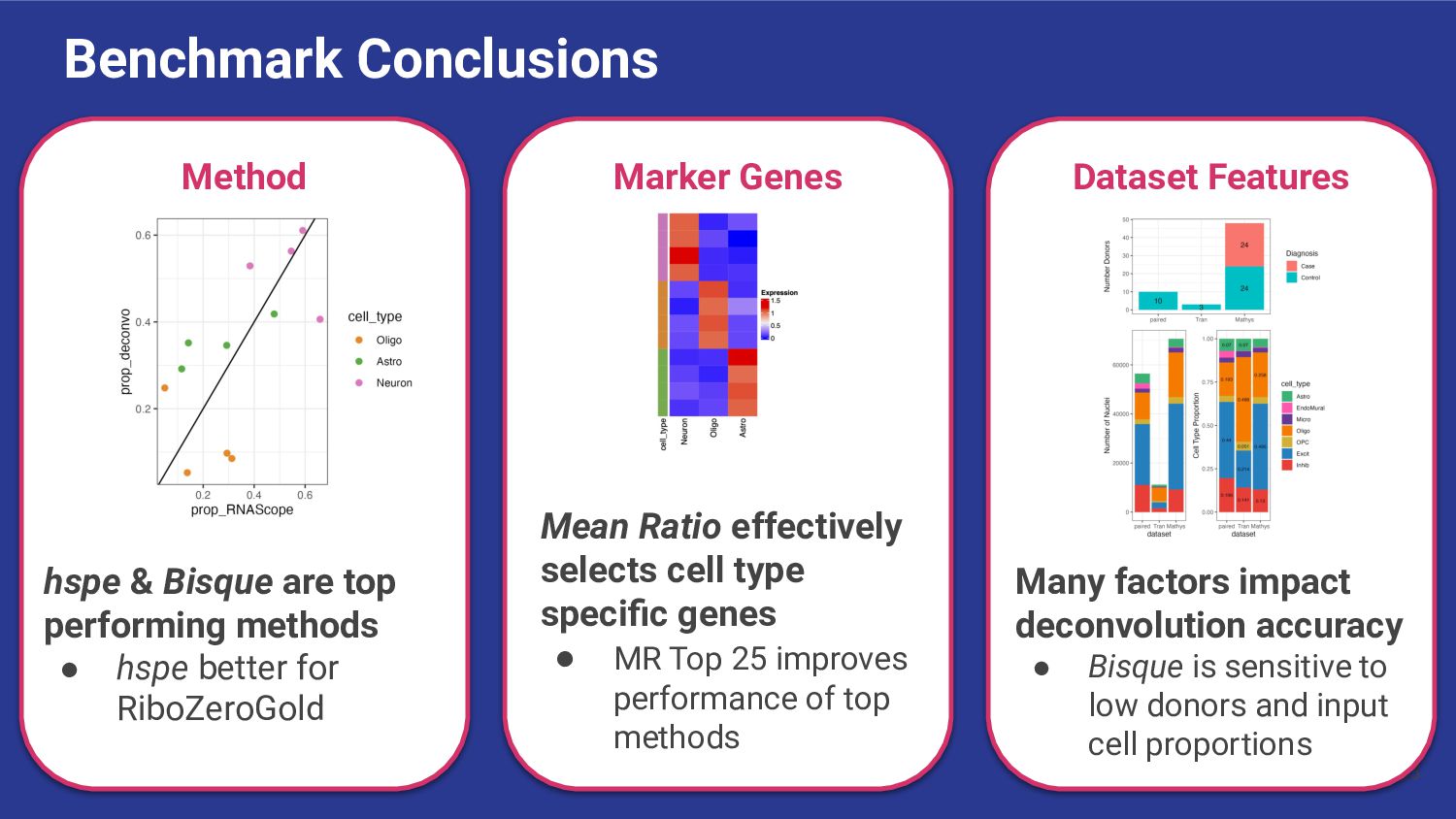
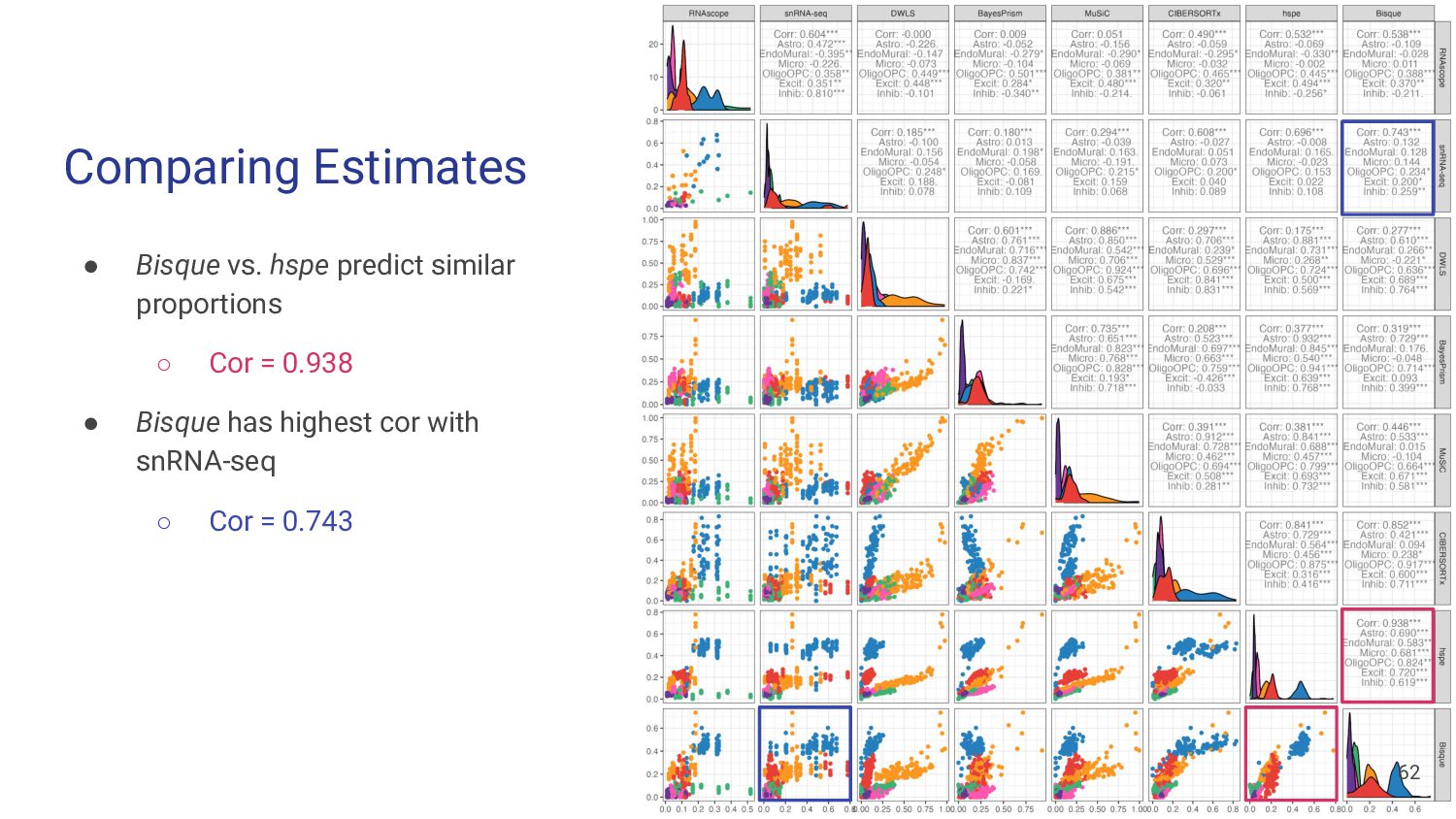
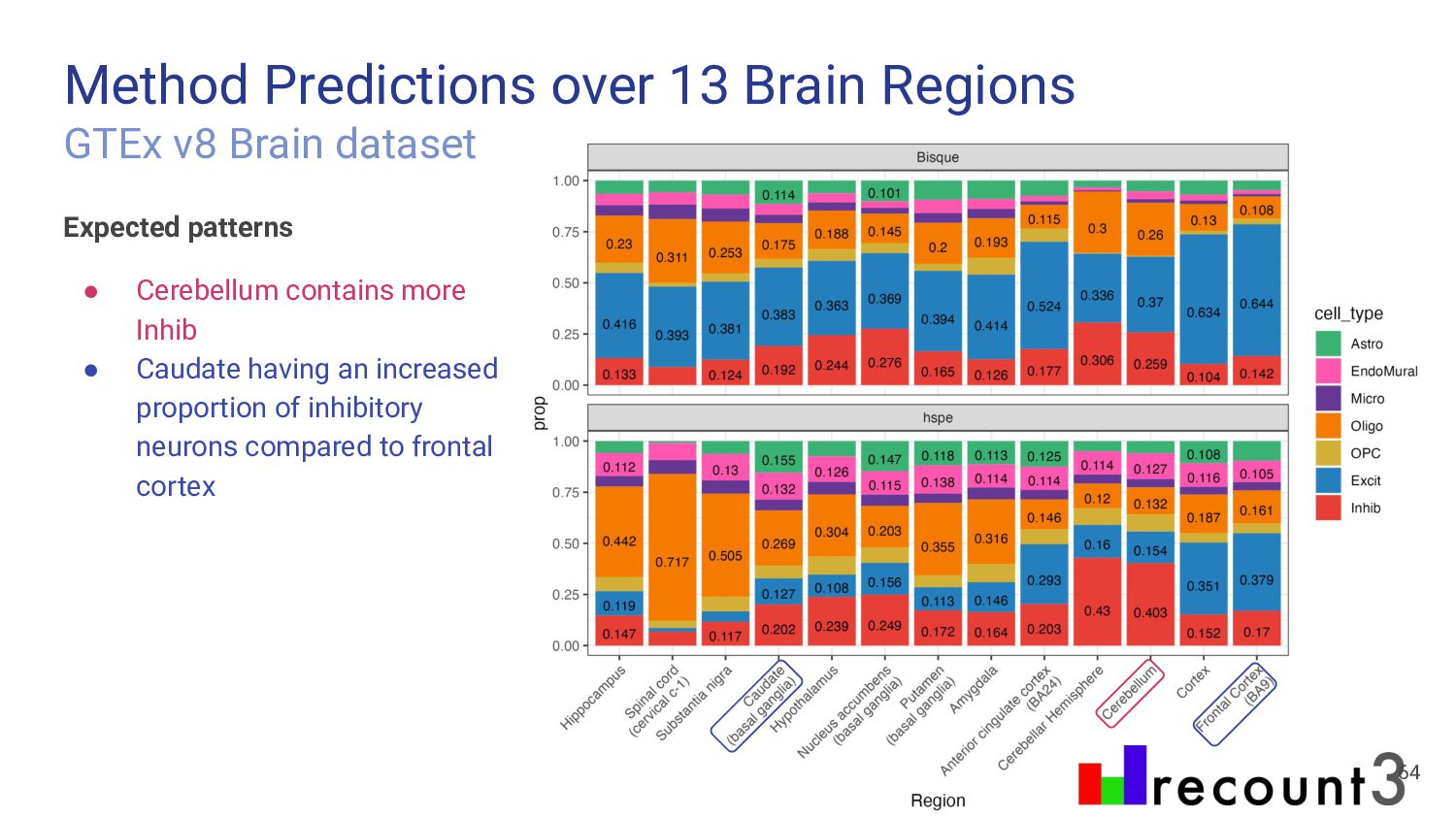
most accurate deconvolution method for brain tissue? hspe & Bisque 2. Is accuracy impacted by type of bulk RNA-seq? Yes a. Library type? Bisque more accurate in polyA, hspe in RiboZeroGold b. RNA extraction? Some impact but inconsistent
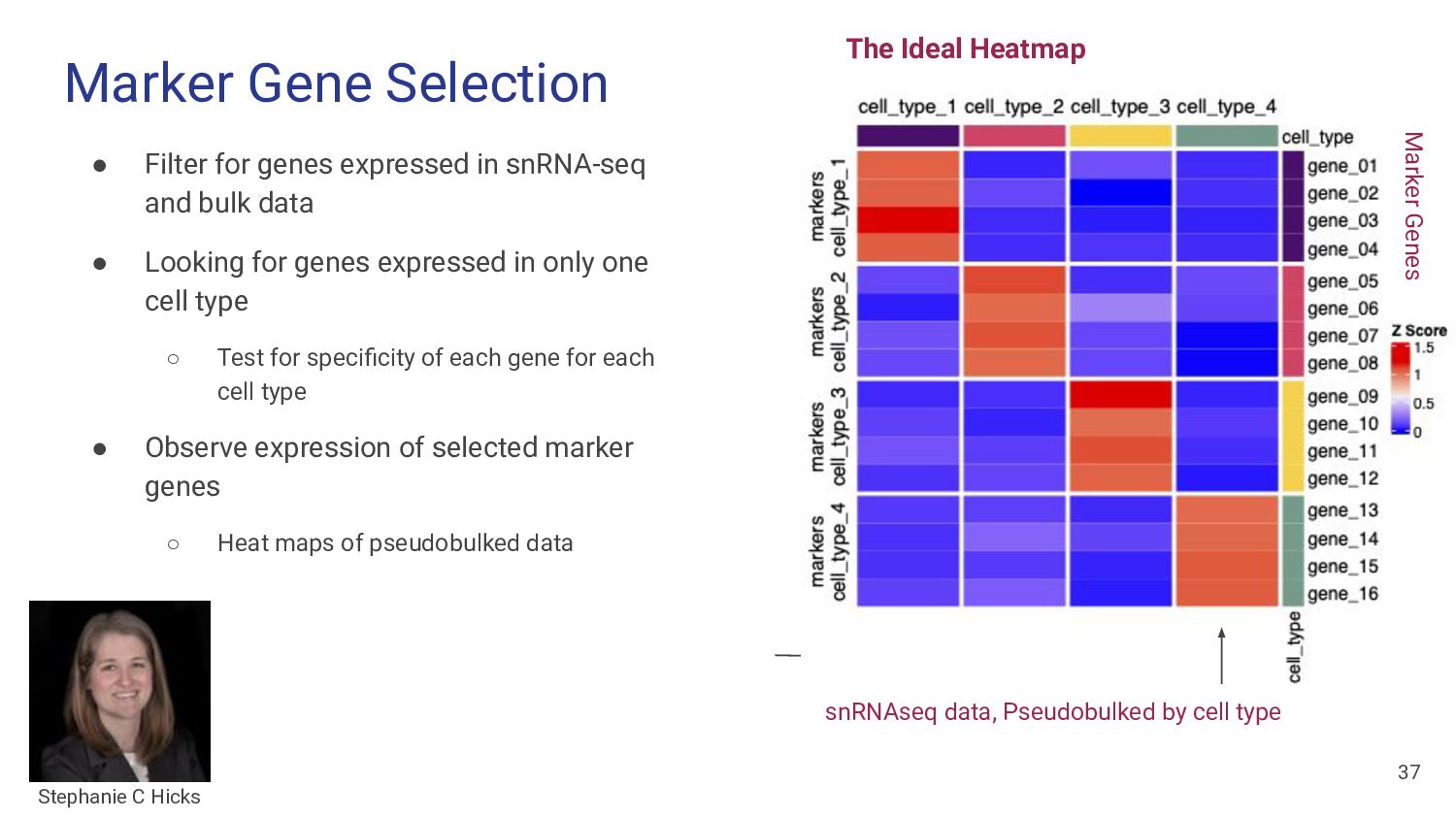
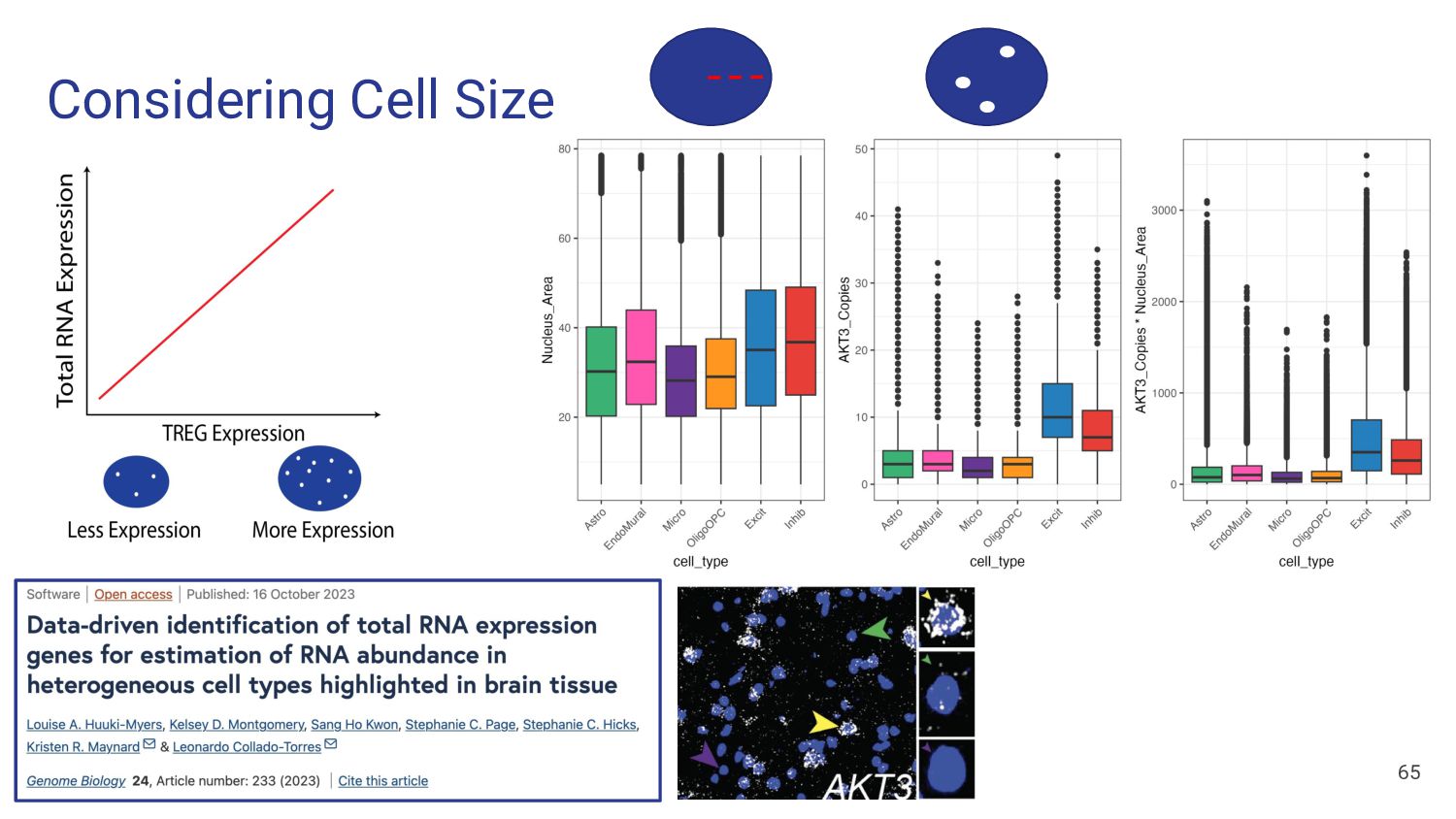
and bulk data • Looking for genes expressed in only one cell type ◦ Test for specificity of each gene for each cell type • Observe expression of selected marker genes ◦ Heat maps of pseudobulked data The Ideal Heatmap snRNAseq data, Pseudobulked by cell type 37 Stephanie C Hicks Marker Genes
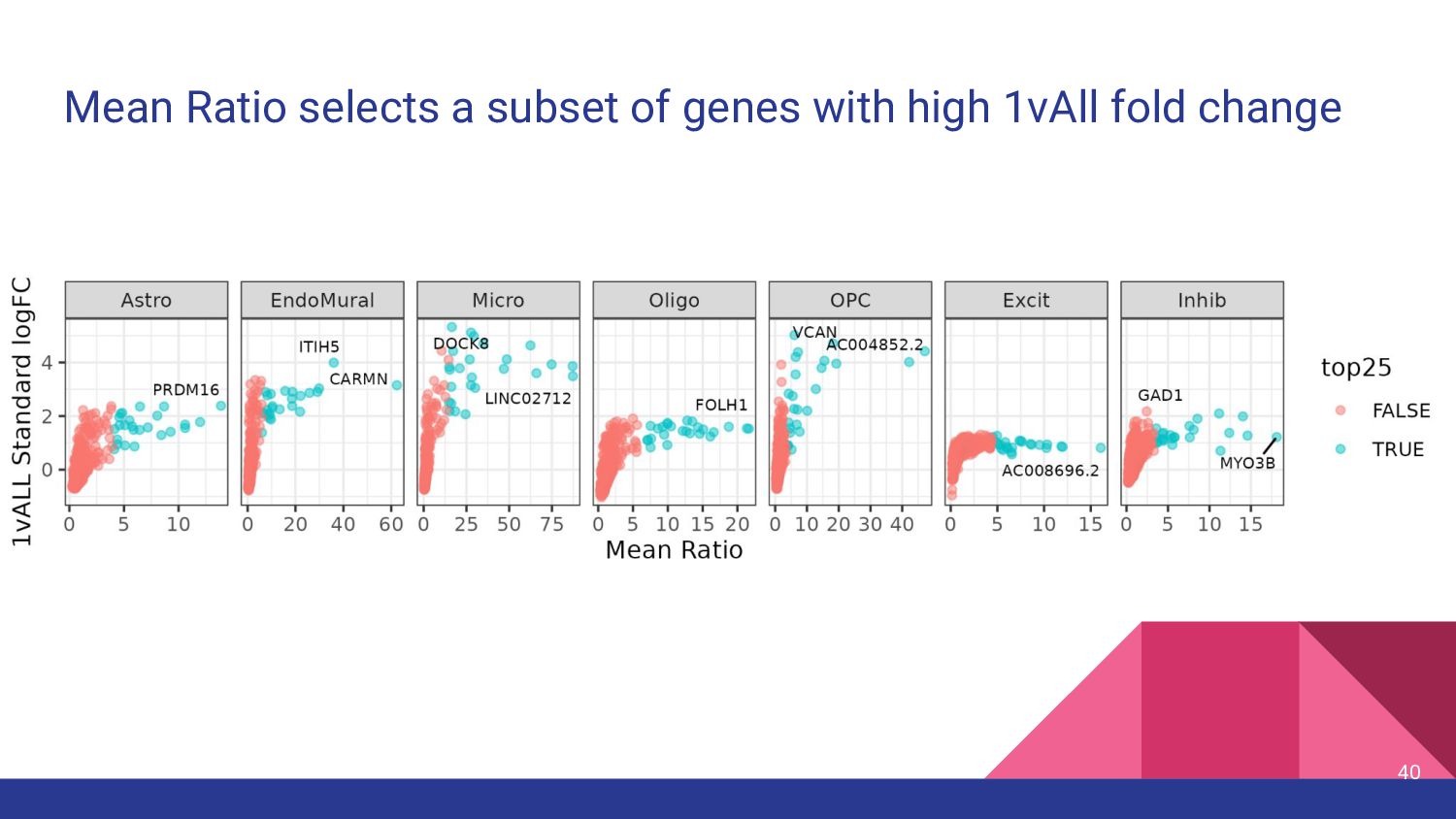
of genes common between the bulk and snRNA-seq datasets 2. 1vALL top25 (145 genes) a. top 25 genes ranked by fold change for each cell type, then filtered to common genes 3. MeanRatio top25 (151 genes) a. top 25 genes ranked by MeanRatio for each cell type, then filtered to common genes 4. MeanRatio over2 (557 genes) a. All genes for each cell type with MeanRatio > 2 5. MeanRatio MAD3 (520 genes) a. All genes for each cell type with MeanRatio > 3 median absolute deviations (MADs) greater than the median of all MeanRatios > 1 41
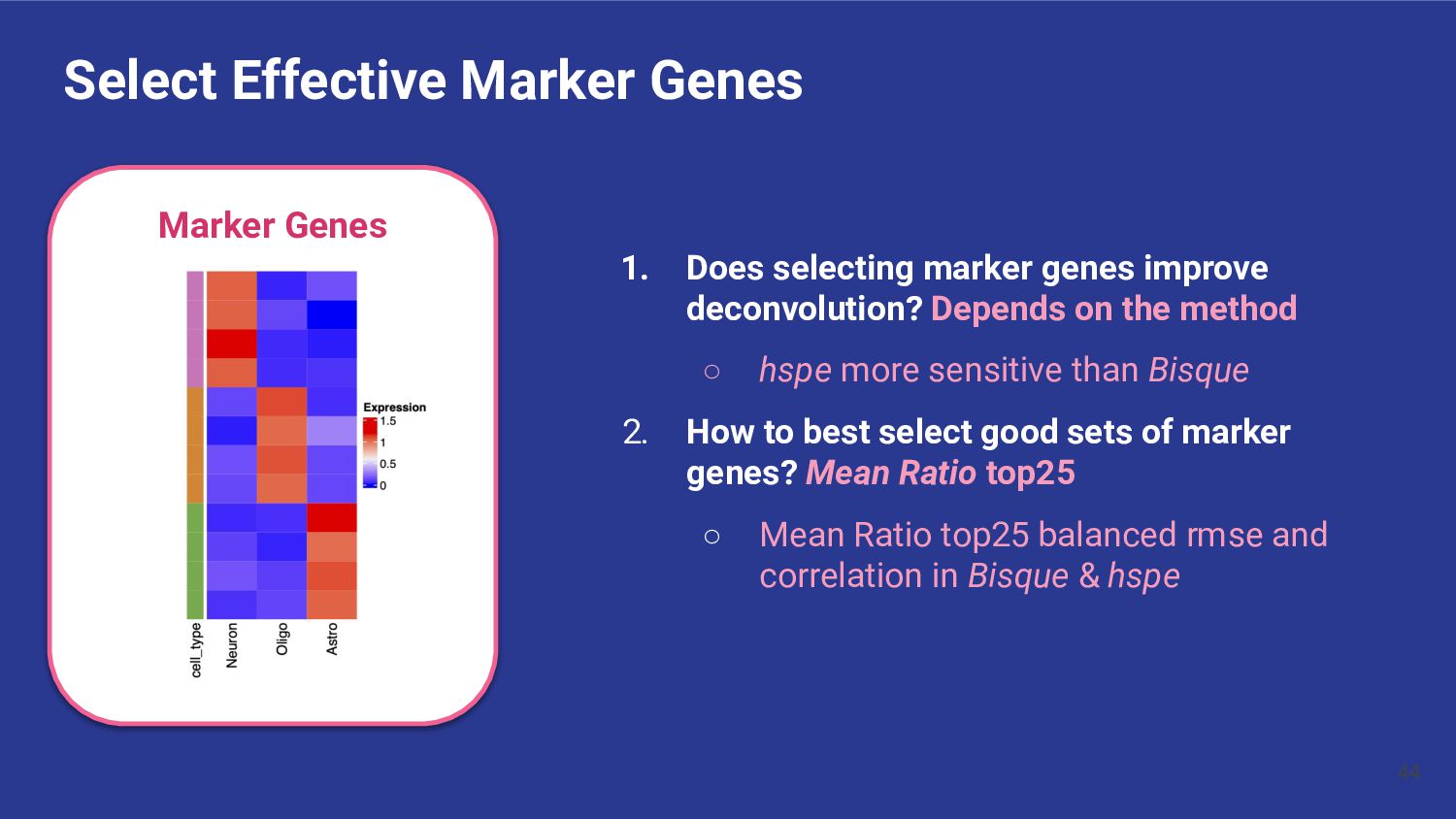
marker genes improve deconvolution? Depends on the method ◦ hspe more sensitive than Bisque 2. How to best select good sets of marker genes? Mean Ratio top25 ◦ Mean Ratio top25 balanced rmse and correlation in Bisque & hspe
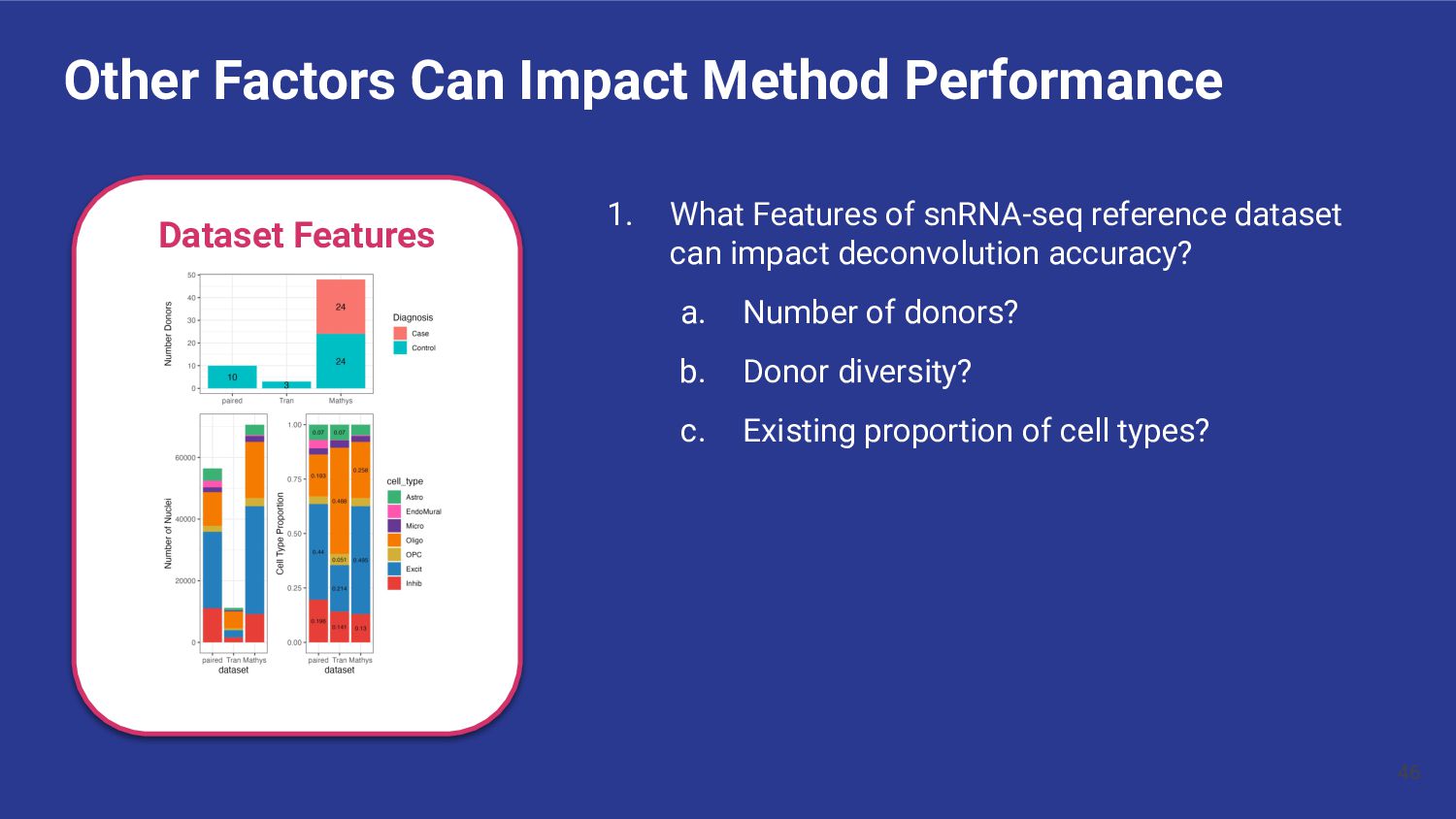
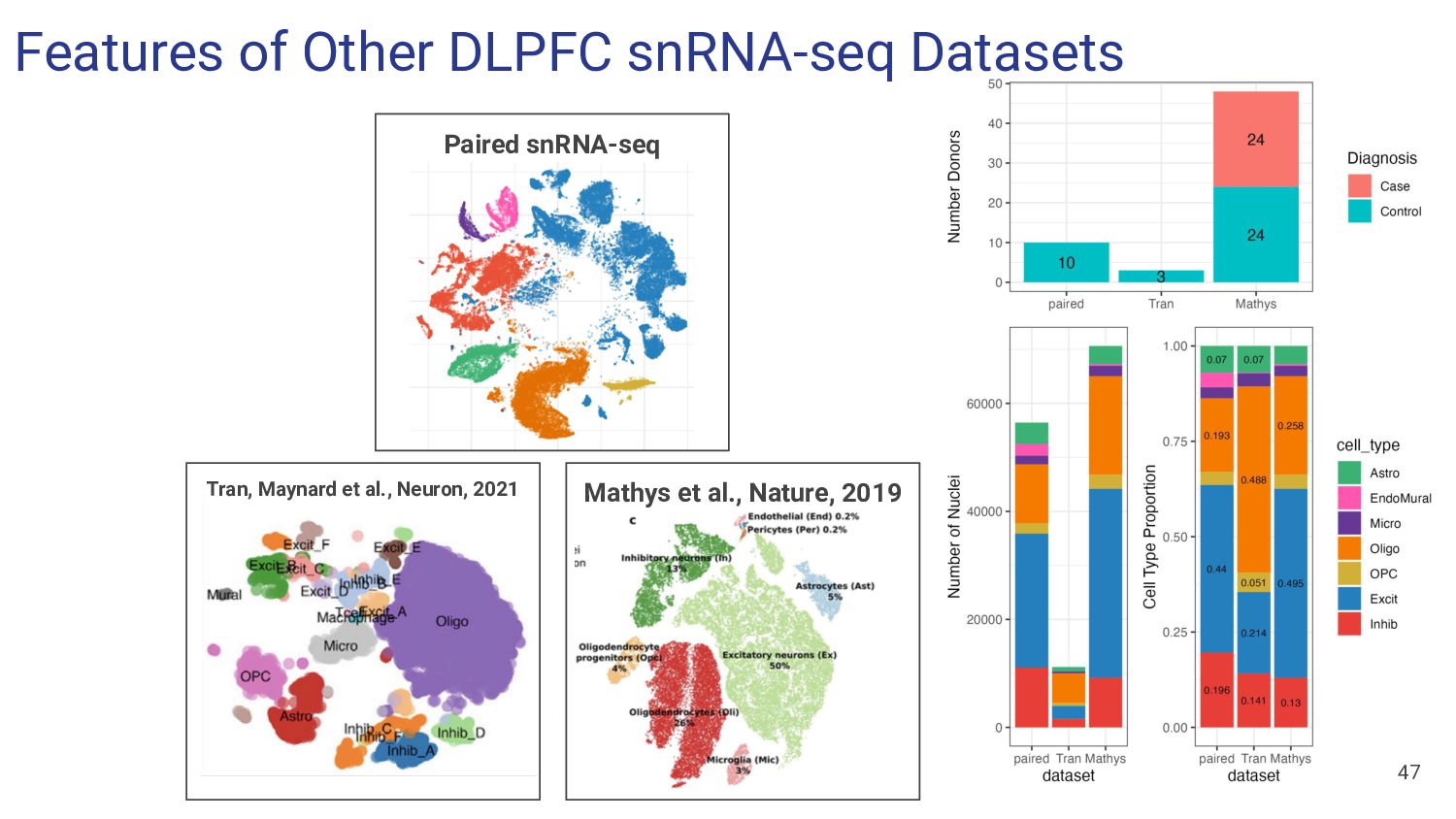
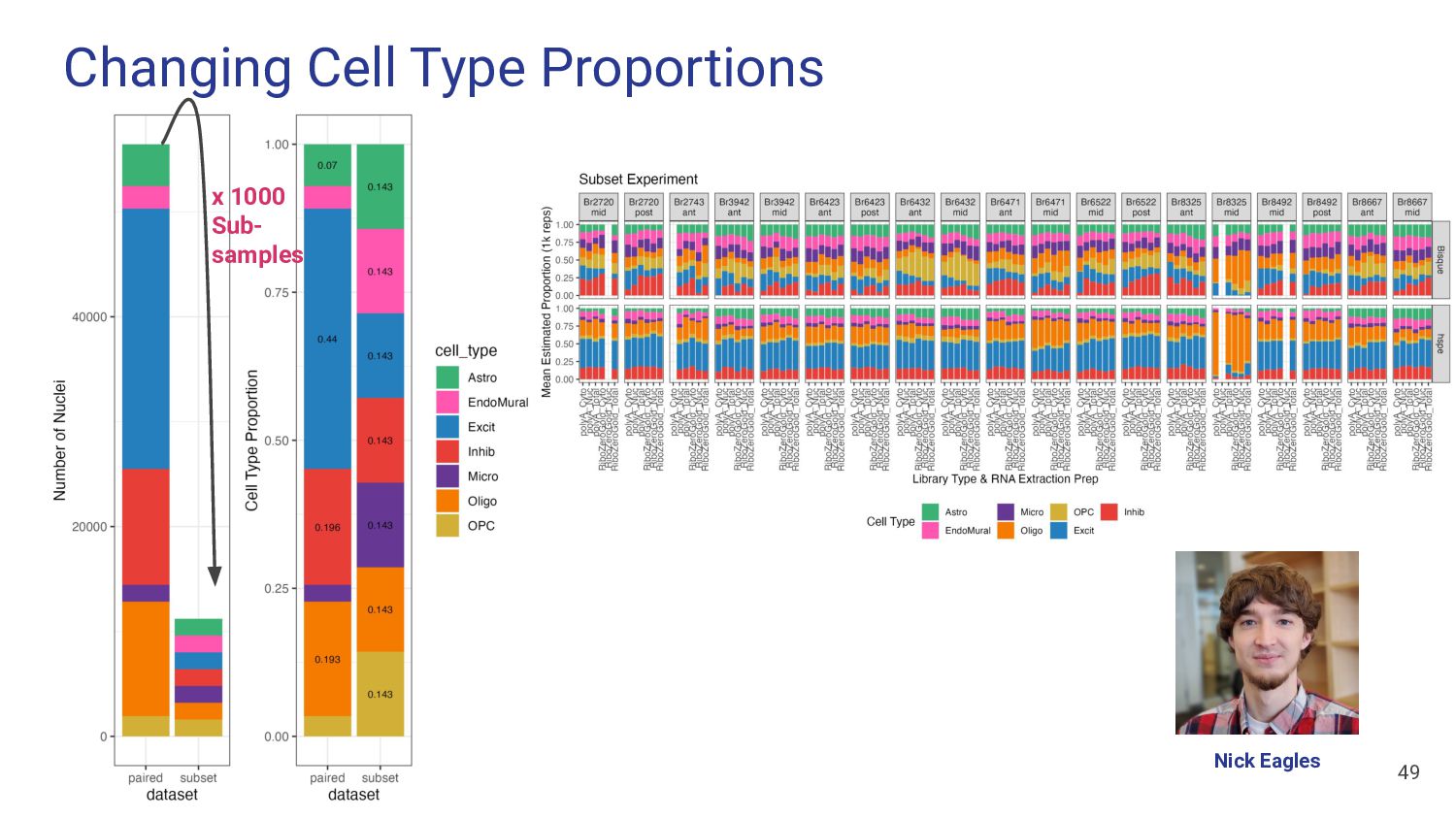
What Features of snRNA-seq reference dataset can impact deconvolution accuracy? a. Number of donors? b. Donor diversity? c. Existing proportion of cell types?
What features of snRNA-seq reference dataset can impact deconvolution accuracy? a. Number of donors? Bisque performs poorly with <4 donors b. Donor diversity? Bisque and hspe were unaffected by inclusion of AD cases c. Existing proportion of cell types? Bisque is biased to snRNA-seq proportions
Bisque are top performing methods • hspe better for RiboZeroGold Mean Ratio effectively selects cell type specific genes • MR Top 25 improves performance of top methods Many factors impact deconvolution accuracy • Bisque is sensitive to low donors and input cell proportions
Strategy Tissue Top Methods Cobos et al. Pseudobulk, Flow sorting Blood, pancreas, kidney DWLS Jin et al. Flow sorting Blood CIBERSORT, MuSiC Dai et al. Immunohistochemistry, scRNA-seq pseudobulk Brain 🧠 dtangle (hspe), Bisque
with tools for marker finding & plotting ◦ github.com/LieberInstitute/DeconvoBuddies ◦ Access paired dataset ▪ Bulk RNA-seq ▪ snRNA-seq data ▪ RNAScope Proportions • Deconvolution code tutorial + video ◦ updated version at LIBD Rstats club on May 3rd 60
Montgomery Sang Ho Kwon Sean Maden Nick Eagles Thank you! Any Questions? Sophia Cinquemani Download these slides: speakerdeck.com/lahuuki @lahuuki Daianna Gonzalez-Padilla NIMH Grant: R01 MH123183 & R01 MH111721
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}