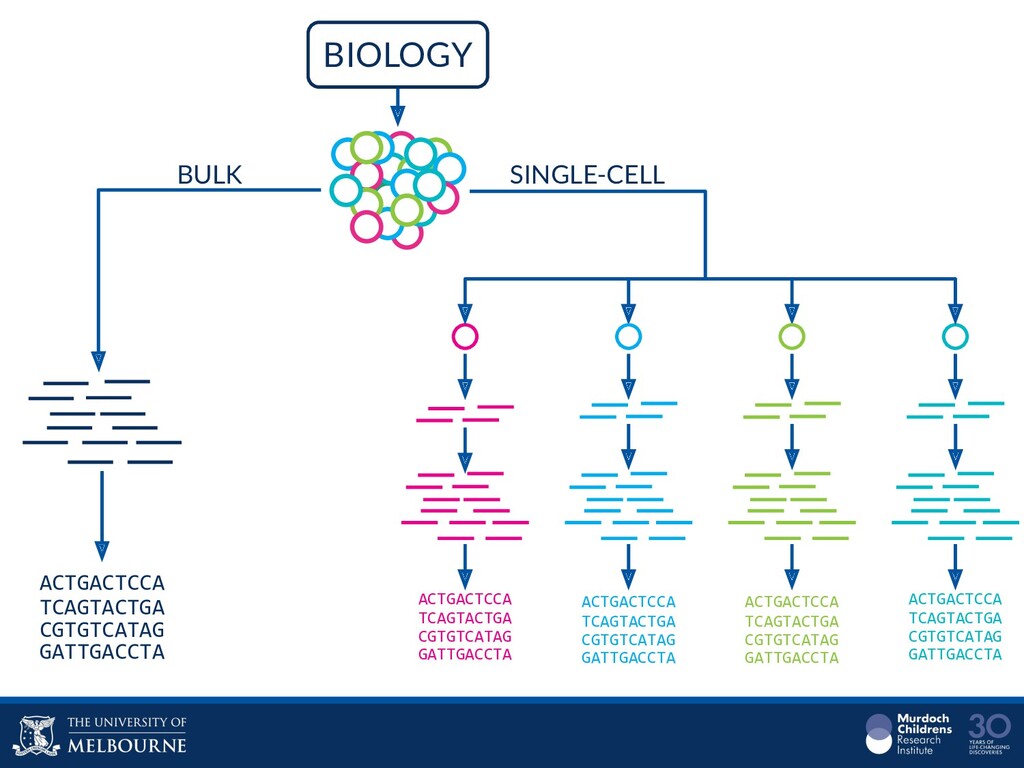
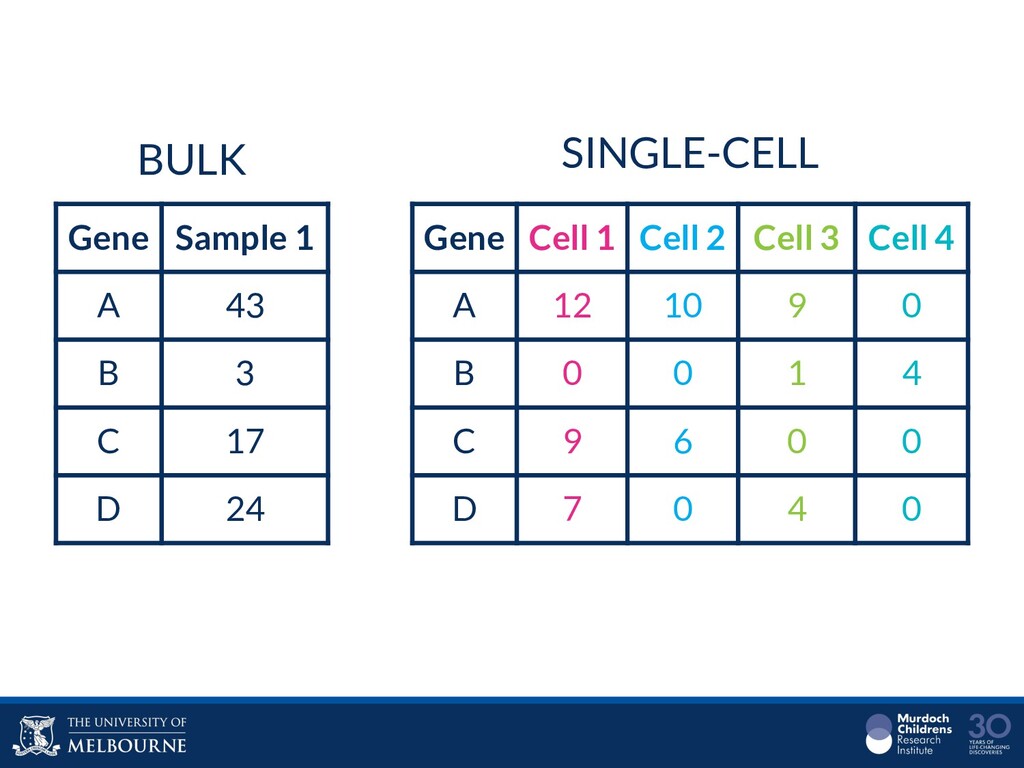
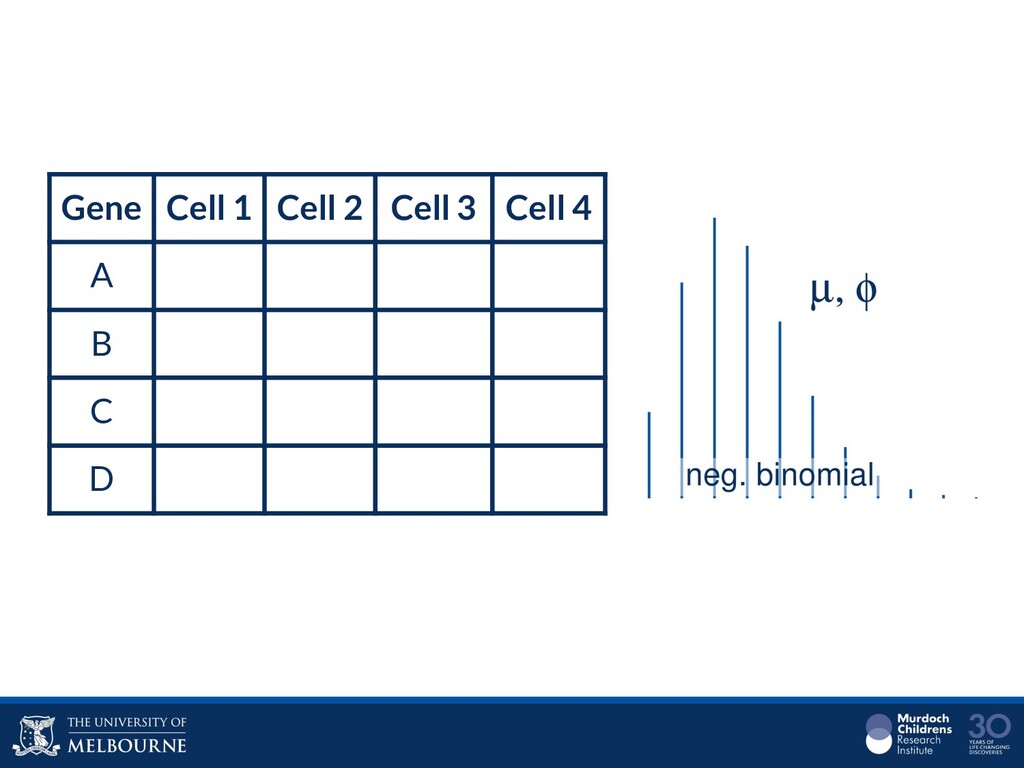
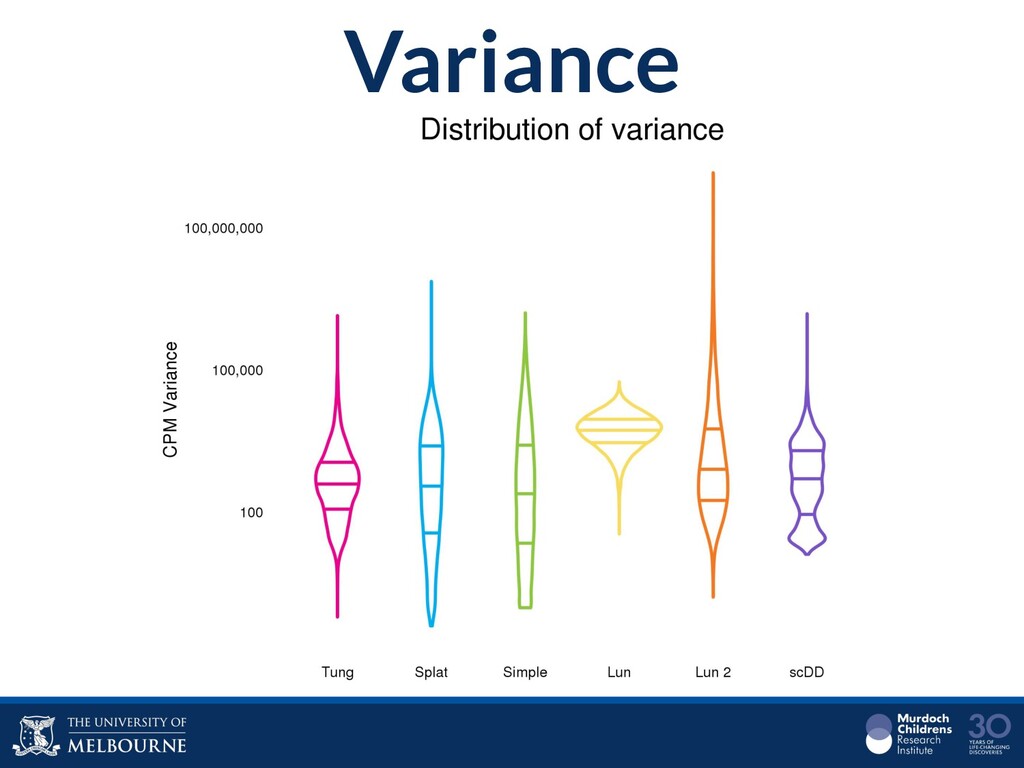
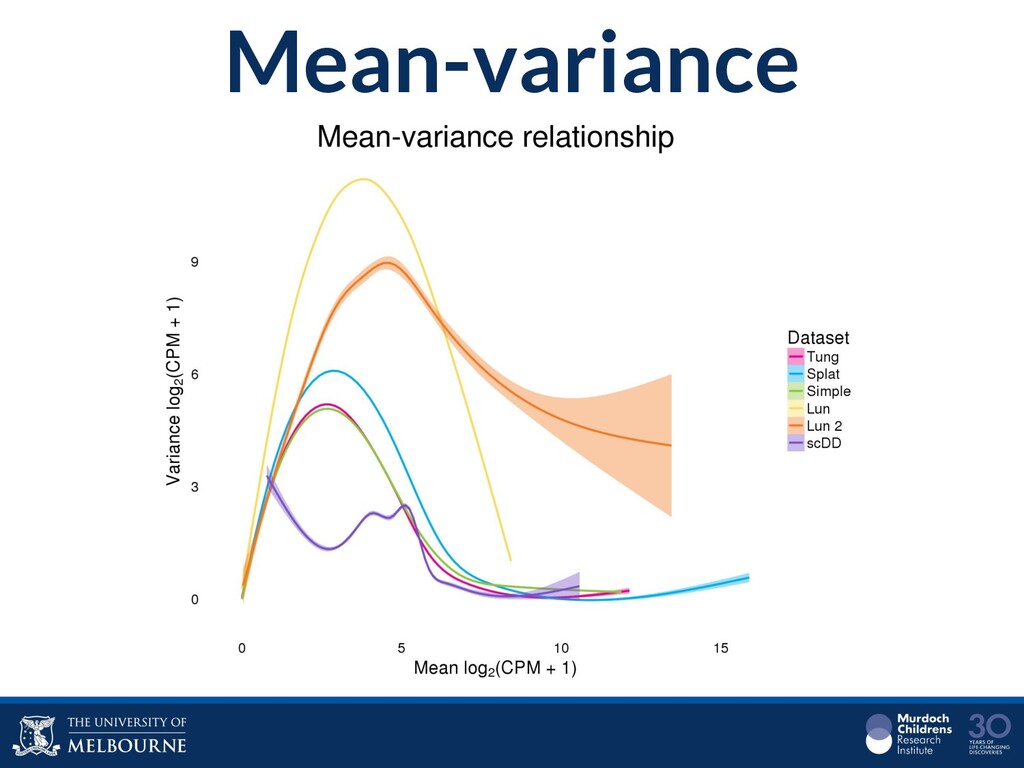
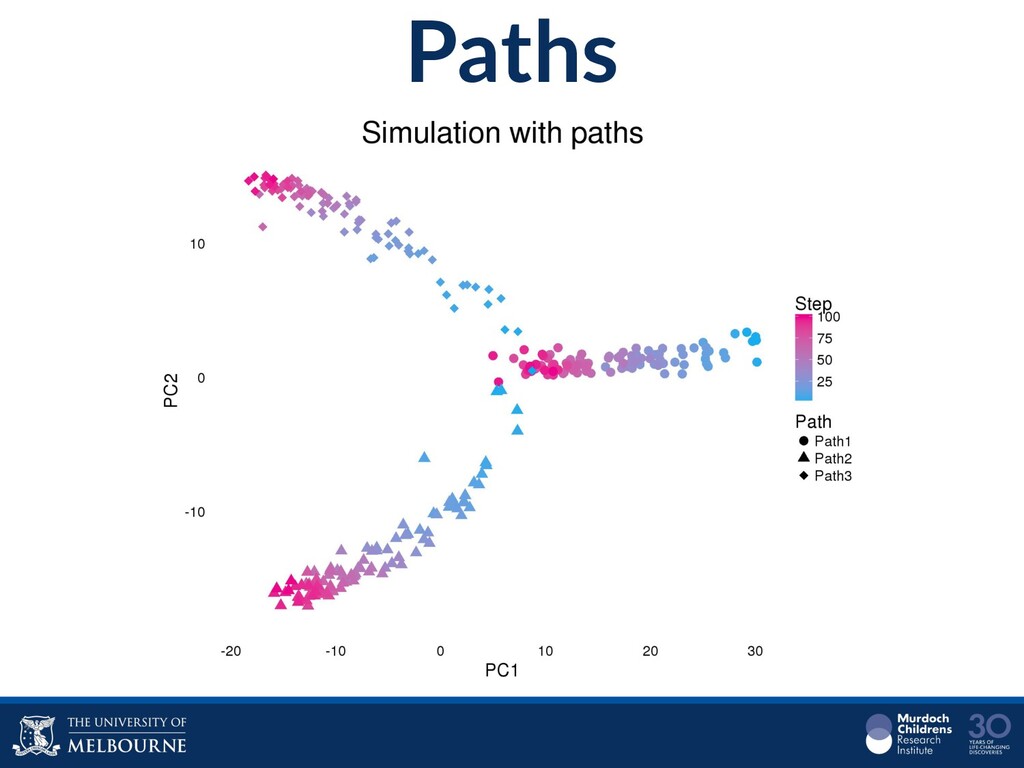
Single-cell RNA sequencing (scRNA-seq) is rapidly becoming a tool of choice for biologists who wish to investigate gene expression, particularly in areas such as development and differentiation. In contrast to traditional bulk RNA-seq experiments, which measure expression averaged across millions of cells, single-cell experiments can be used to observe how genes are expressed in individual cells. Along with the dramatic increase in resolution provided by scRNA-seq comes an array of bioinformatics challenges. Single-cell data is relatively sparse (for both biological and technical reasons), quality control is difficult and it is unclear how to replicate measurements. The focus of analysis is also different, with more emphasis on clustering cells to identify cell types or ordering of cells to understand dynamic processes than traditional tasks such as differential expression testing. Any new bioinformatics method for scRNA-seq analysis should demonstrate two things: 1) it can do what it claims and 2) it helps to produce biological insight. The first is hard to prove on real data where there is often no known truth. Because of this, bioinformaticians turn to simulations. Unfortunately current scRNA-seq simulations are frequently poorly documented, not reproducible and do not demonstrate similarity to real data or experimental designs. Here we discuss some of the problems with simulating scRNA-seq data and provide a simulation framework that addresses these concerns.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}