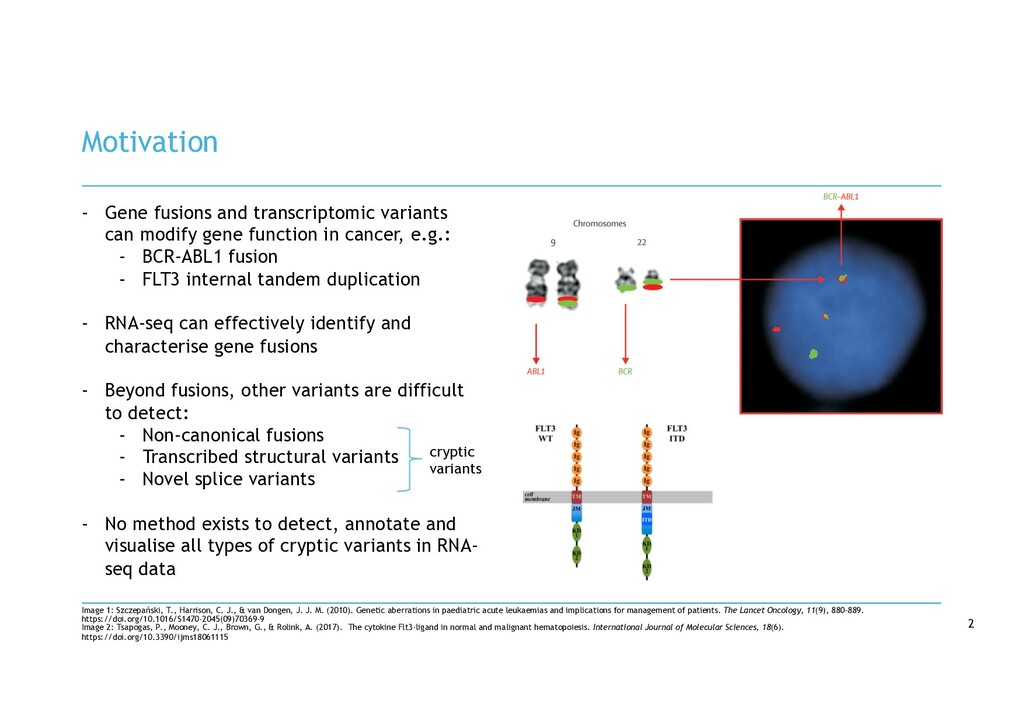
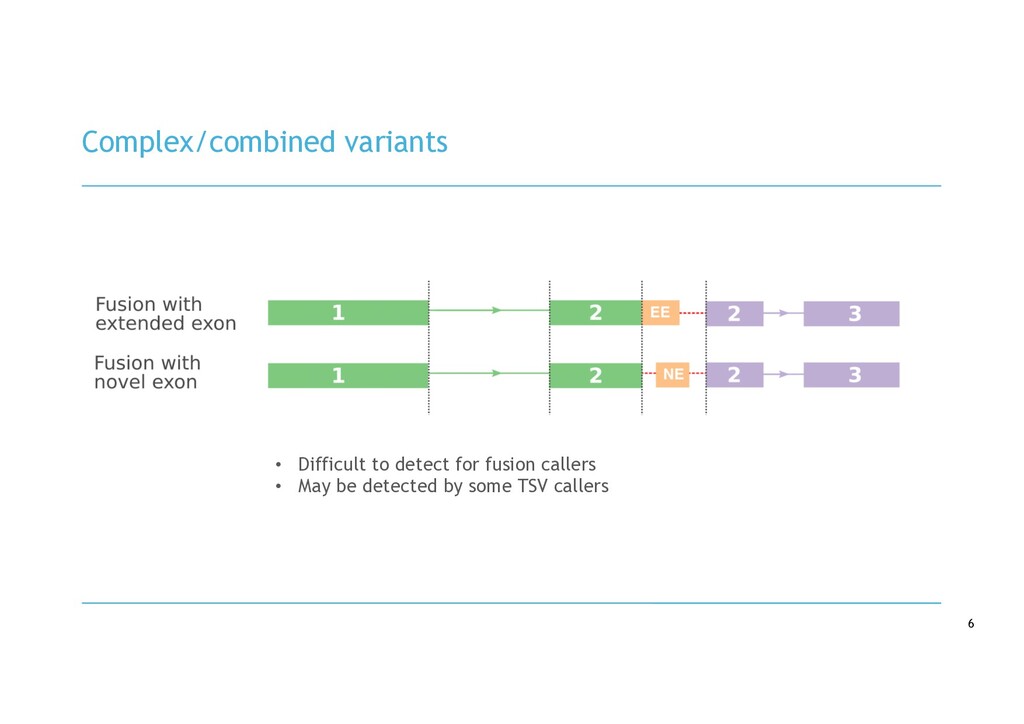
van Dongen, J. J. M. (2010). Genetic aberrations in paediatric acute leukaemias and implications for management of patients. The Lancet Oncology, 11(9), 880–889. https://doi.org/10.1016/S1470-2045(09)70369-9 Image 2: Tsapogas, P., Mooney, C. J., Brown, G., & Rolink, A. (2017). The cytokine Flt3-ligand in normal and malignant hematopoiesis. International Journal of Molecular Sciences, 18(6). https://doi.org/10.3390/ijms18061115 - Gene fusions and transcriptomic variants can modify gene function in cancer, e.g.: - BCR-ABL1 fusion - FLT3 internal tandem duplication - RNA-seq can effectively identify and characterise gene fusions - Beyond fusions, other variants are difficult to detect: - Non-canonical fusions - Transcribed structural variants - Novel splice variants - No method exists to detect, annotate and visualise all types of cryptic variants in RNA- seq data cryptic variants
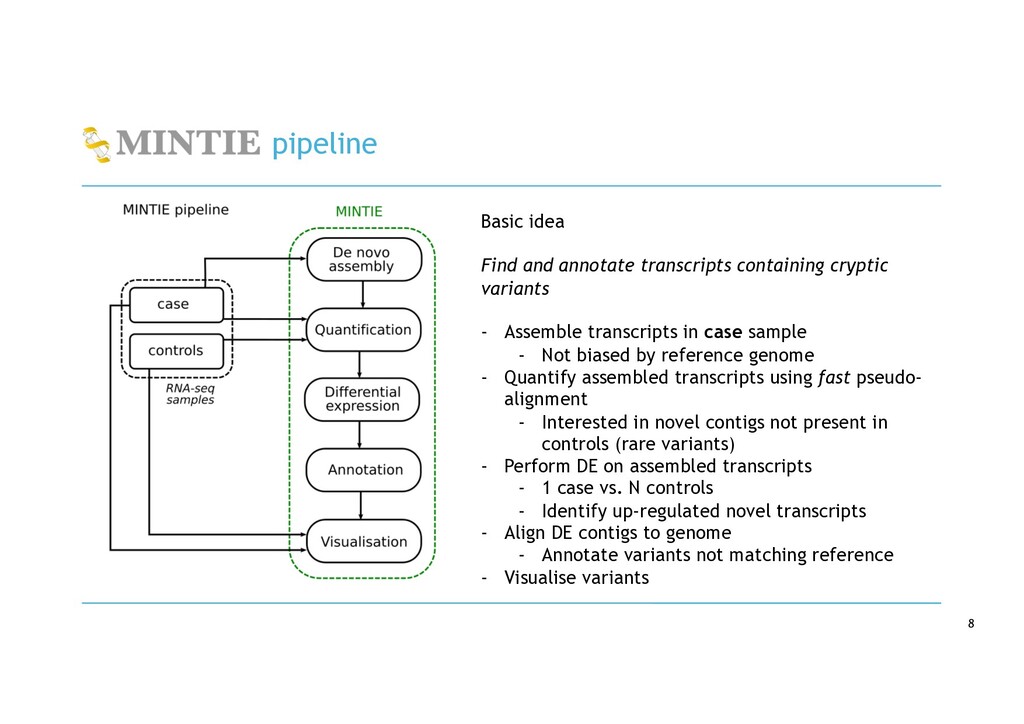
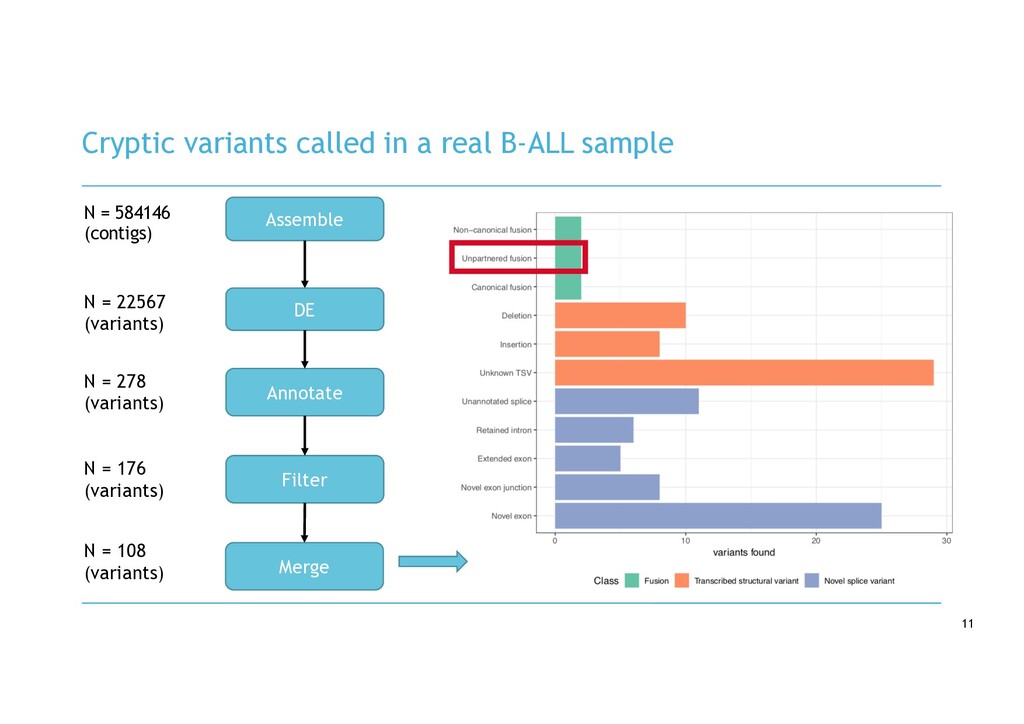
variants - Assemble transcripts in case sample - Not biased by reference genome - Quantify assembled transcripts using fast pseudo- alignment - Interested in novel contigs not present in controls (rare variants) - Perform DE on assembled transcripts - 1 case vs. N controls - Identify up-regulated novel transcripts - Align DE contigs to genome - Annotate variants not matching reference - Visualise variants
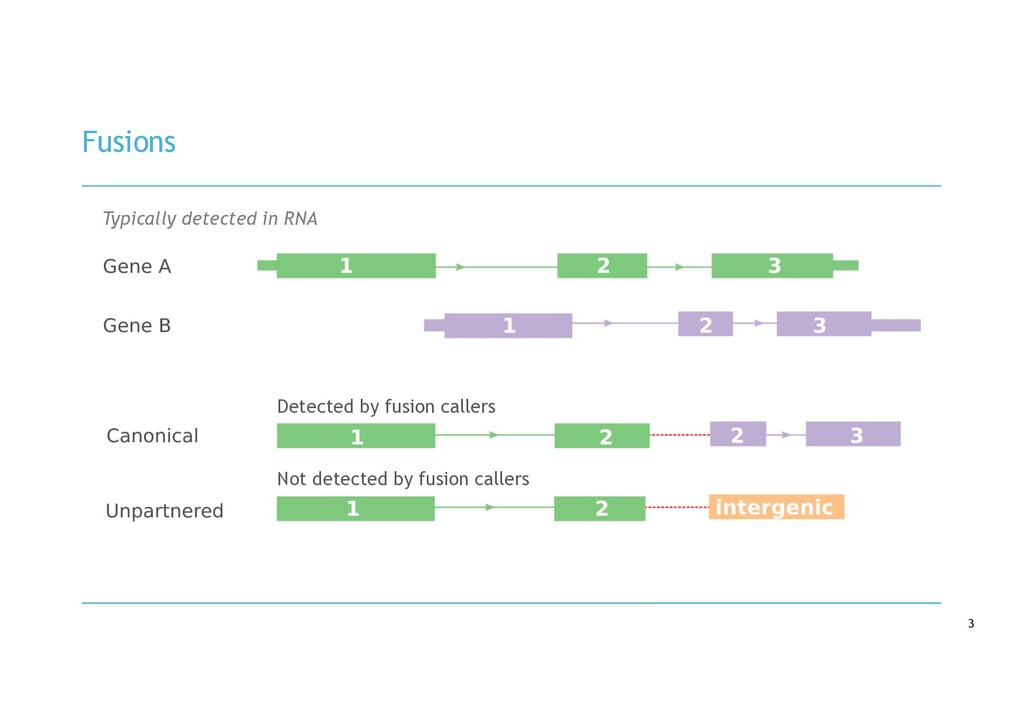
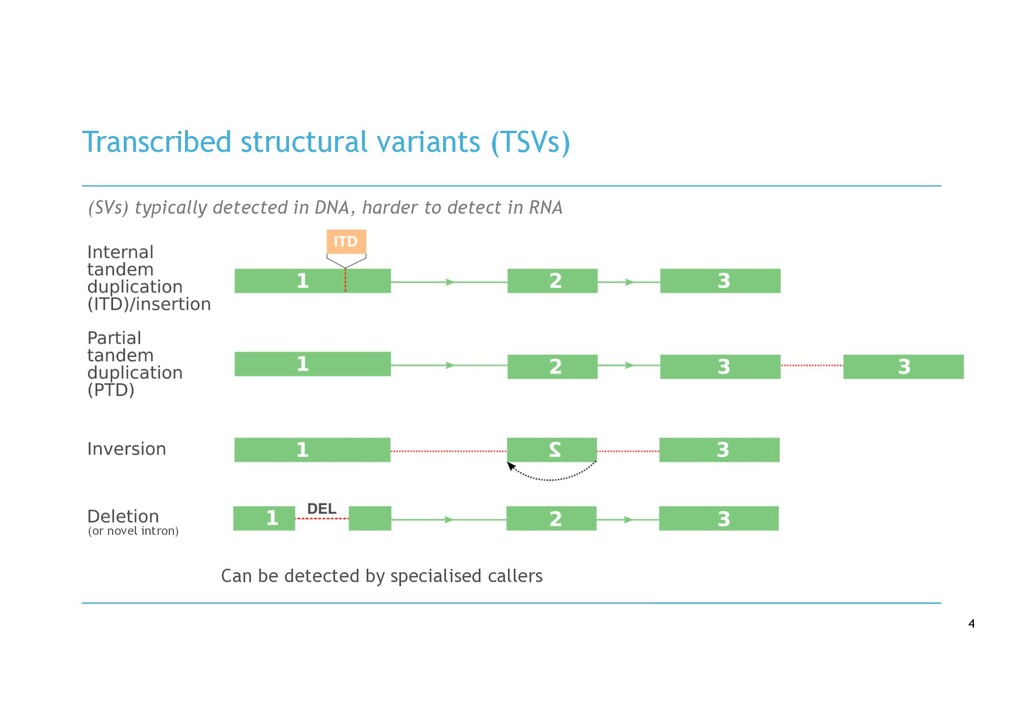
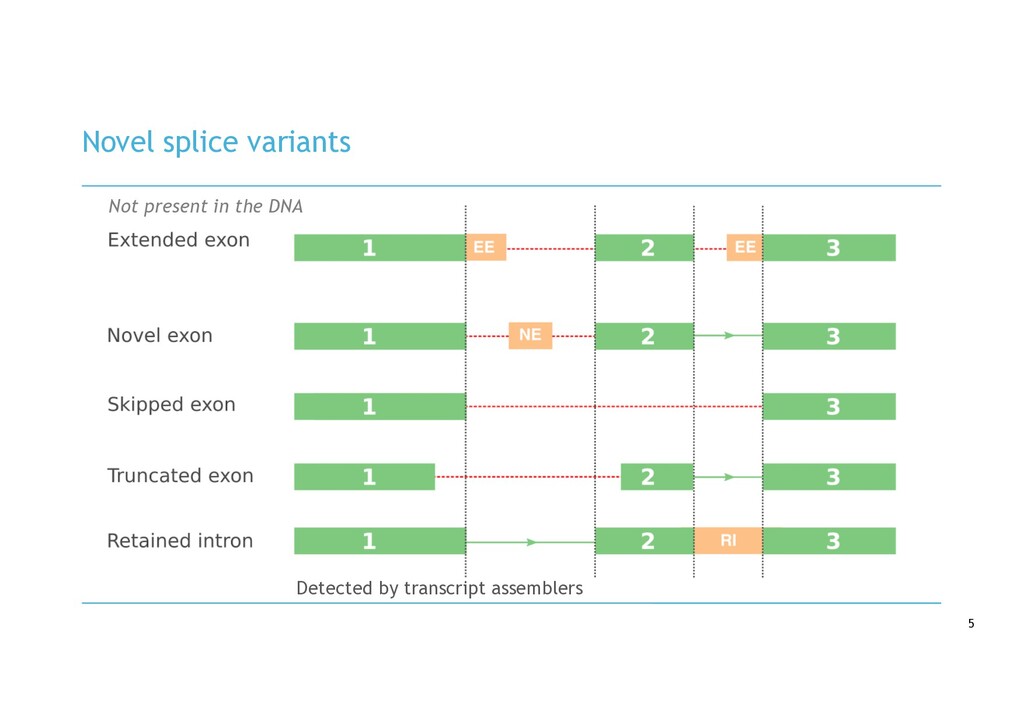
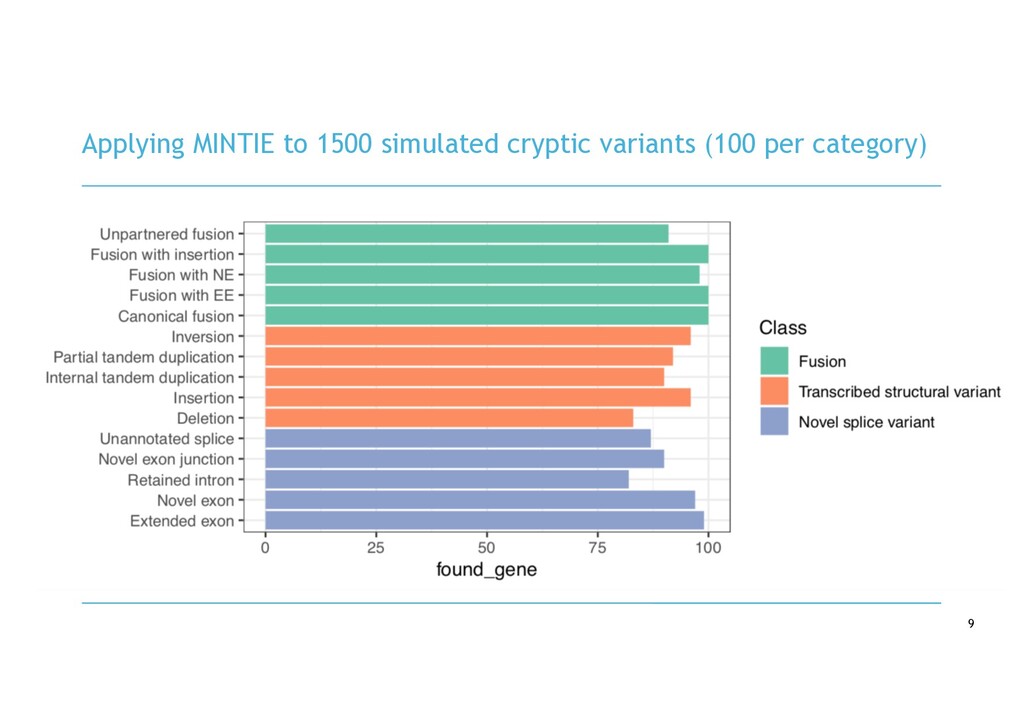
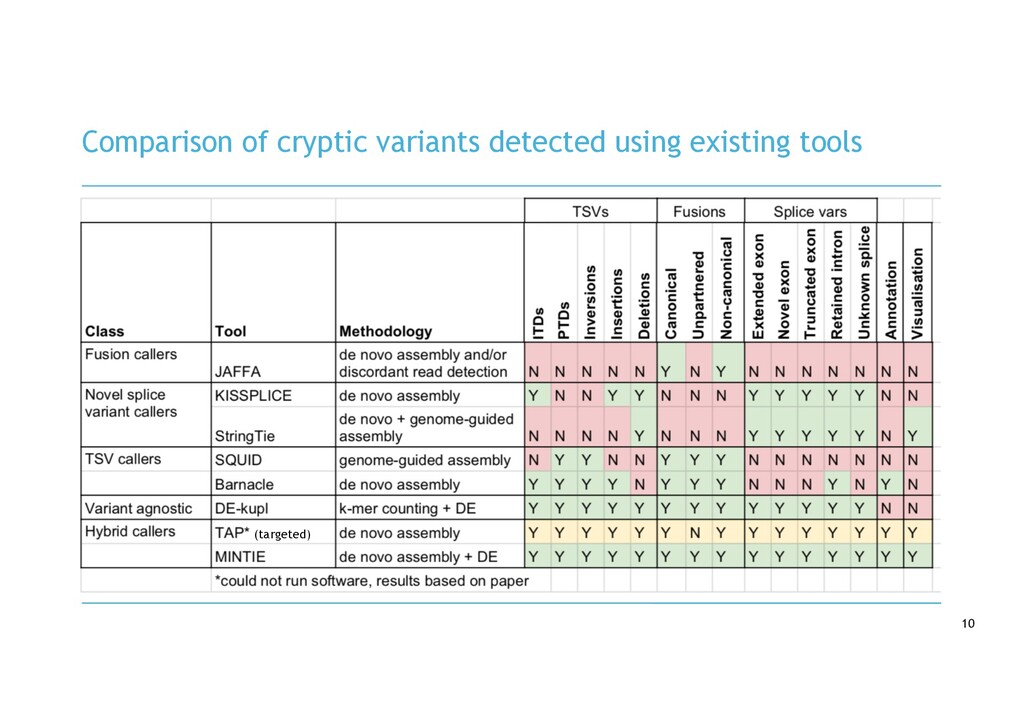
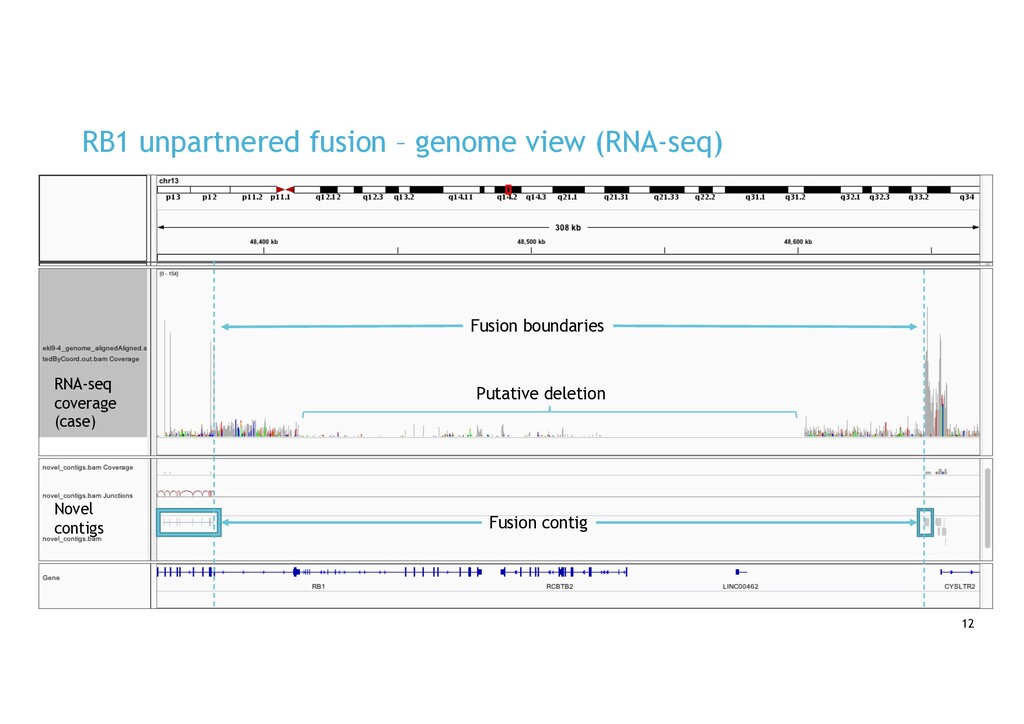
in RNA-seq cancer samples: • Canonical, non-canonical and unpartnered fusions • Novel splice variants • Transcribed structural variants • Method • De novo assemble > quantify > DE > annotate > visualise • Detects more variants than any other tool • Detected RB1 unpartnered fusion and IKZF1 PTD in B-ALL samples • We are hard at work on the visualisation component!
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}