Spectra: Why? Some motivations… Aid in the identification of fundamental vibrational modes (e.g. no overtones, leakage, artifacts…); Identification of active modes too weak to be accurately positioned; Identification of the number of modes involved in relatively broad bands; Reliable starting frequencies for deconvolution of experimental spectra. Determination of spectra of crystals having a perfectly defined composition: end members of solid solutions; solid solutions having a specific particular chemistry; no impurities. Raman or IR band assignment to vibrations of specific groups or atoms; Correlation of spectra of similar substances. The first bold statement…
presentation: A very brief and condensed presentation of the theory; The application to an archeologically interesting material: jadeite (jade); The correlation of the vibrational features of jadeite with other structurally similar phases having a different composition (omphacite and diopside) What You will see here...
) ( ) , ( R x R E R x H Hamiltonian Operator Wave Function, depending upon the electronic spatial and spin coordinates (x) and nuclear (R) Energy of the system (parametrically depending upon R) ) , ( s r x r: space coordinates; s: spin coordinates of all the electrons A Brief Look at the Theory The key for the calculation of vibrational spectra (and a lot of other properties) is in the solution of the time-independent Schroedinger equation, within the limit of the Born-Oppenheimer approximation (separability of electronic and nuclear motions)
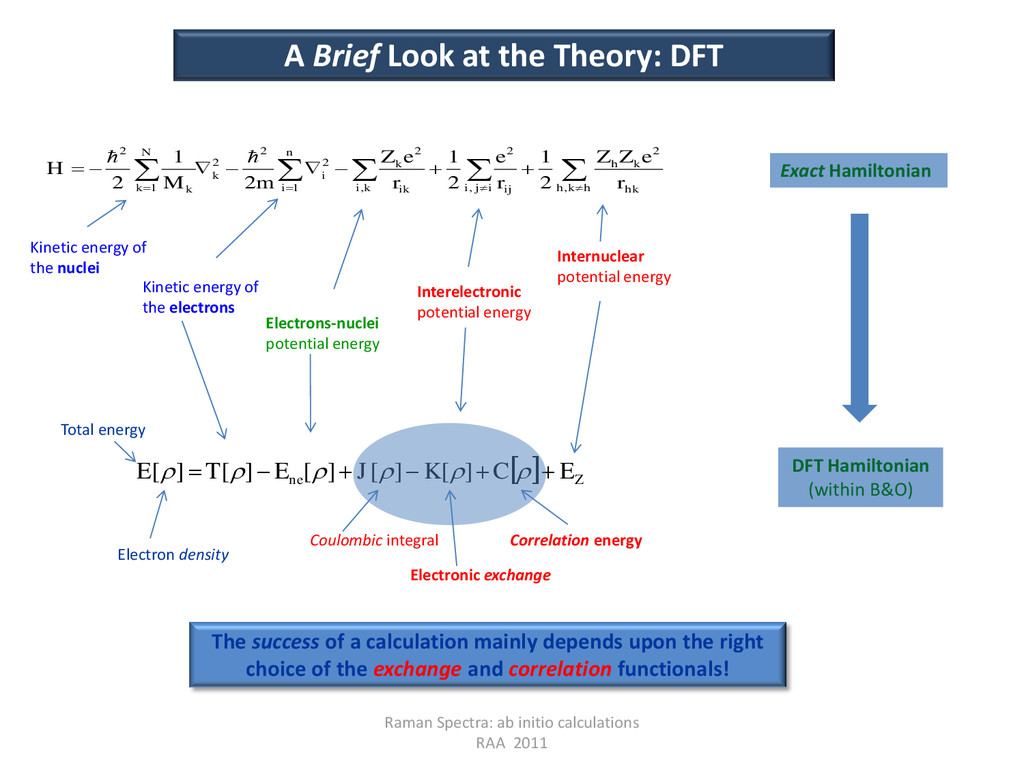
at the Theory: DFT Exact Hamiltonian DFT Hamiltonian (within B&O) k i i j i h k h hk k h ij ik k n i i k N k k r e Z Z r e r e Z m M H , , , 2 2 2 1 2 2 2 1 2 2 1 2 1 2 1 2 Kinetic energy of the nuclei Kinetic energy of the electrons Electrons-nuclei potential energy Interelectronic potential energy Internuclear potential energy Z ne E C K J E T E ] [ ] [ ] [ ] [ ] [ Electron density Total energy Coulombic integral Electronic exchange Correlation energy The success of a calculation mainly depends upon the right choice of the exchange and correlation functionals!
q q E E q q q E R E 3 , 2 3 2 1 2 1 ) 0 ( ) , , , ( ) ( The effective potential E(R) which rules the nuclear motion, can be expanded in a Taylor’s series of some mass-weighted coordinates q; by truncating the series at the second order in the q’s (harmonic approximation), and by taking into account that at the equilibrium (E/qj )=0, we get: i i i i i i i Q h Q Q H i ) ( 2 1 2 1 2 2 2 2 2 2 2 1 2 1 ) ( Q Q Q h ) ( ) ( , , , i i m i m i i m i Q Q h i i i i m h m 2 1 , ) 2 1 ( , By representing the Hamiltonian (H) in the space of the q’s, and diagonalizing, the wave function (), describing the nuclear motion, is factorized in the product of 1D-functions (normal modes) i i i m N m Q Q Q i ) ( ) , , ( , 3 1 Raman Spectra: ab initio calculations RAA 2011 A Brief Look at the Theory: Vibrations
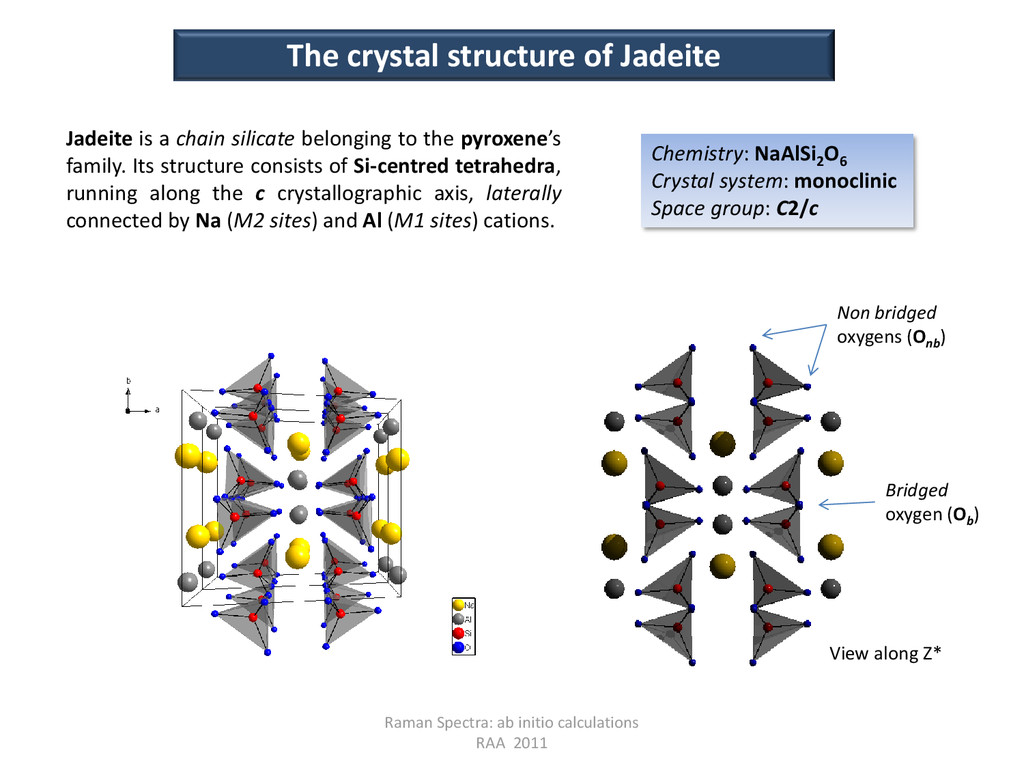
of Jadeite Jadeite is a chain silicate belonging to the pyroxene’s family. Its structure consists of Si-centred tetrahedra, running along the c crystallographic axis, laterally connected by Na (M2 sites) and Al (M1 sites) cations. Bridged oxygen (Ob ) Non bridged oxygens (Onb ) Chemistry: NaAlSi2 O6 Crystal system: monoclinic Space group: C2/c View along Z*
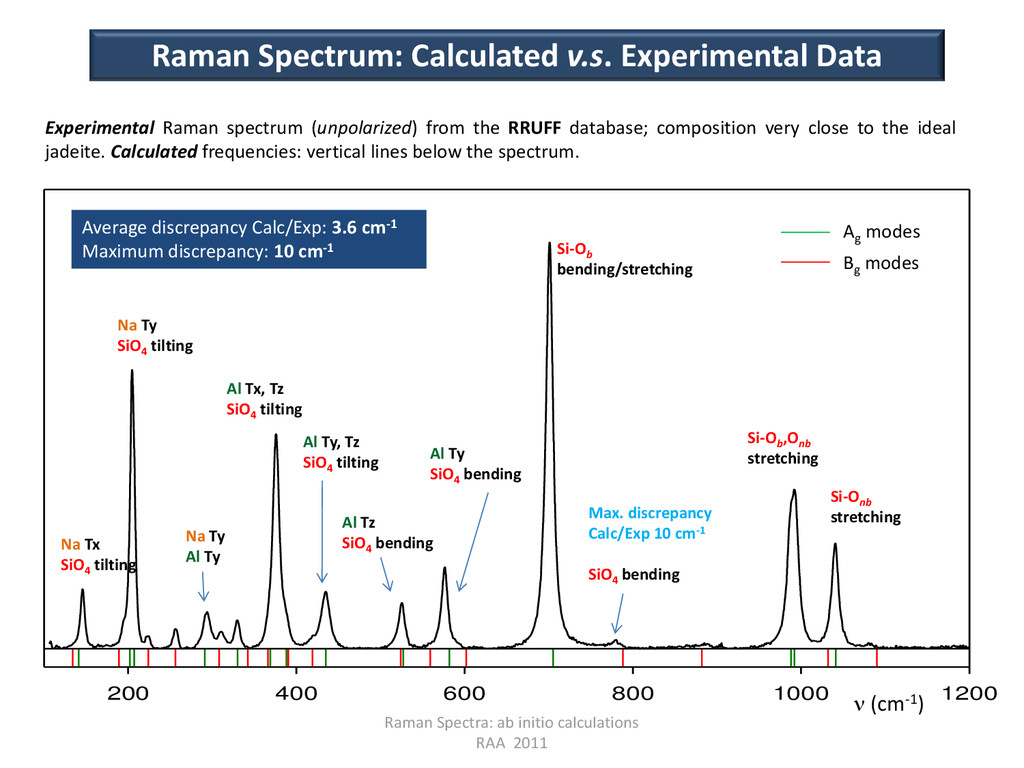
v.s. Experimental Data Experimental Raman spectrum (unpolarized) from the RRUFF database; composition very close to the ideal jadeite. Calculated frequencies: vertical lines below the spectrum. 200 400 600 800 1000 1200 Ag modes Bg modes Si-Ob bending/stretching Si-Onb stretching Si-Ob ,Onb stretching Al Ty SiO4 bending Al Tz SiO4 bending Al Ty, Tz SiO4 tilting Al Tx, Tz SiO4 tilting Na Ty SiO4 tilting Na Tx SiO4 tilting Na Ty Al Ty Max. discrepancy Calc/Exp 10 cm-1 SiO4 bending Average discrepancy Calc/Exp: 3.6 cm-1 Maximum discrepancy: 10 cm-1 (cm-1)
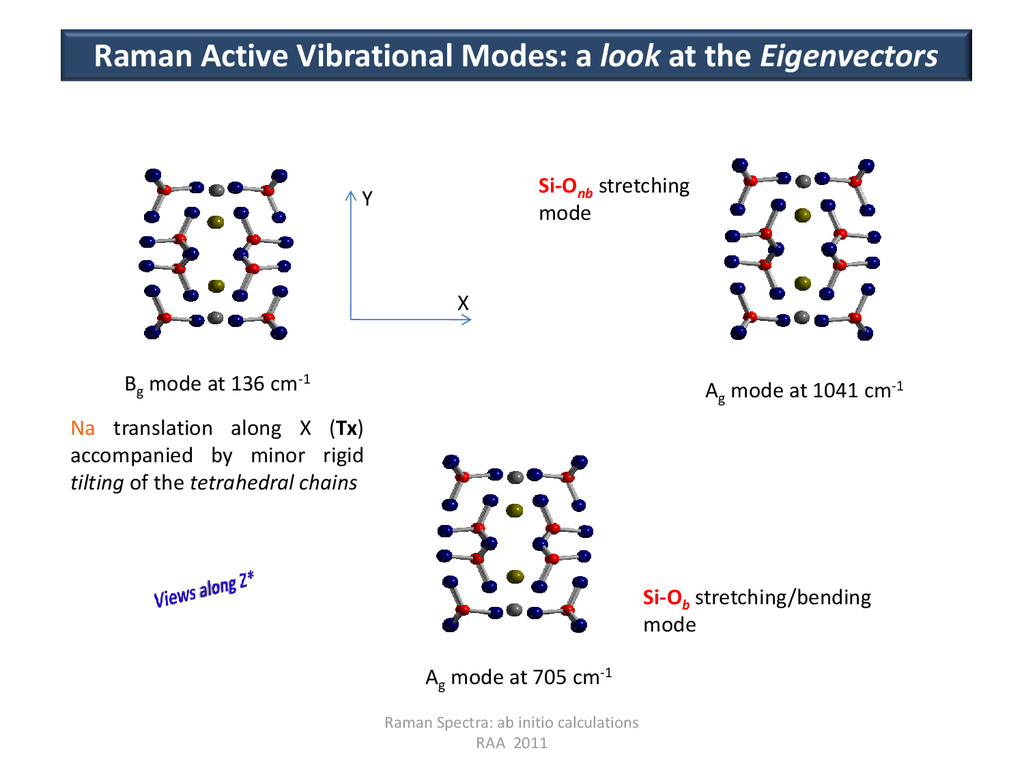
Modes: a look at the Eigenvectors Bg mode at 136 cm-1 Ag mode at 705 cm-1 Ag mode at 1041 cm-1 X Y Na translation along X (Tx) accompanied by minor rigid tilting of the tetrahedral chains Si-Ob stretching/bending mode Si-Onb stretching mode
-1/4 -1/4 -1/4 -1/4 a p b p a c b c X Y Na atom b* C Raman Spectra: ab initio calculations RAA 2011 Phonon Dispersion in Jadeite Space Group C2/c 2 possible unit cells: (i) a primitive (P) cell with (ap , bp ) base vectors; (ii) a centered (C) cell with (ac , bc ) base vectors. In reciprocal space, the point Y = 1/2(a* P +b* P ) is at a special position of the Brillouin zone (BZ), along the [110]* P direction, and corresponds to the (010)C reciprocal lattice vector. Therefore, at Y, the modulation wave of the lattice vibrations is directed along [010]C and has a periodicity corresponding to the length of the vector bc At Y, the Na atoms having x=1/2 will move in antiphase with respect to the Na atoms at x=0 (or 1) Section of the reciprocal lattice. Zone axis: [001] a* C Γ a* P b* P (110)P Y Λ BZ b* C
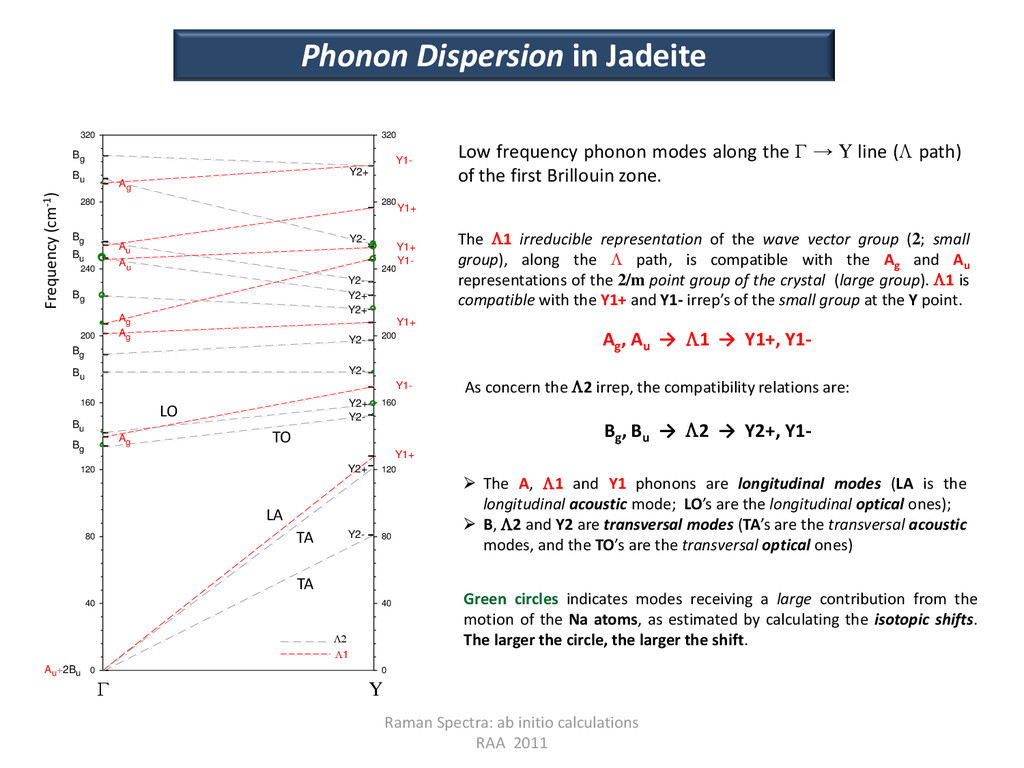
40 80 120 160 200 240 280 320 Y2- Y2+ Y1+ Au+2Bu Bg Ag Bu Y2- Y2+ Y1- Y2- Y2- Y1+ Y2+ Y2+ Y2- Y1- Y1+ Y2- Y1+ Y2+ Y1- 1 Bu Bg Ag Ag Bg Au Bu Au Bg Ag Bu Bg Raman Spectra: ab initio calculations RAA 2011 Γ Y Frequency (cm-1) Low frequency phonon modes along the Γ → Y line ( path) of the first Brillouin zone. The 1 irreducible representation of the wave vector group (2; small group), along the path, is compatible with the Ag and Au representations of the 2/m point group of the crystal (large group). 1 is compatible with the Y1+ and Y1- irrep’s of the small group at the Y point. Ag , Au → 1 → Y1+, Y1- As concern the 2 irrep, the compatibility relations are: Bg , Bu → 2 → Y2+, Y1- The A, 1 and Y1 phonons are longitudinal modes (LA is the longitudinal acoustic mode; LO’s are the longitudinal optical ones); B, 2 and Y2 are transversal modes (TA’s are the transversal acoustic modes, and the TO’s are the transversal optical ones) LA TA TA TO LO Green circles indicates modes receiving a large contribution from the motion of the Na atoms, as estimated by calculating the isotopic shifts. The larger the circle, the larger the shift. Phonon Dispersion in Jadeite
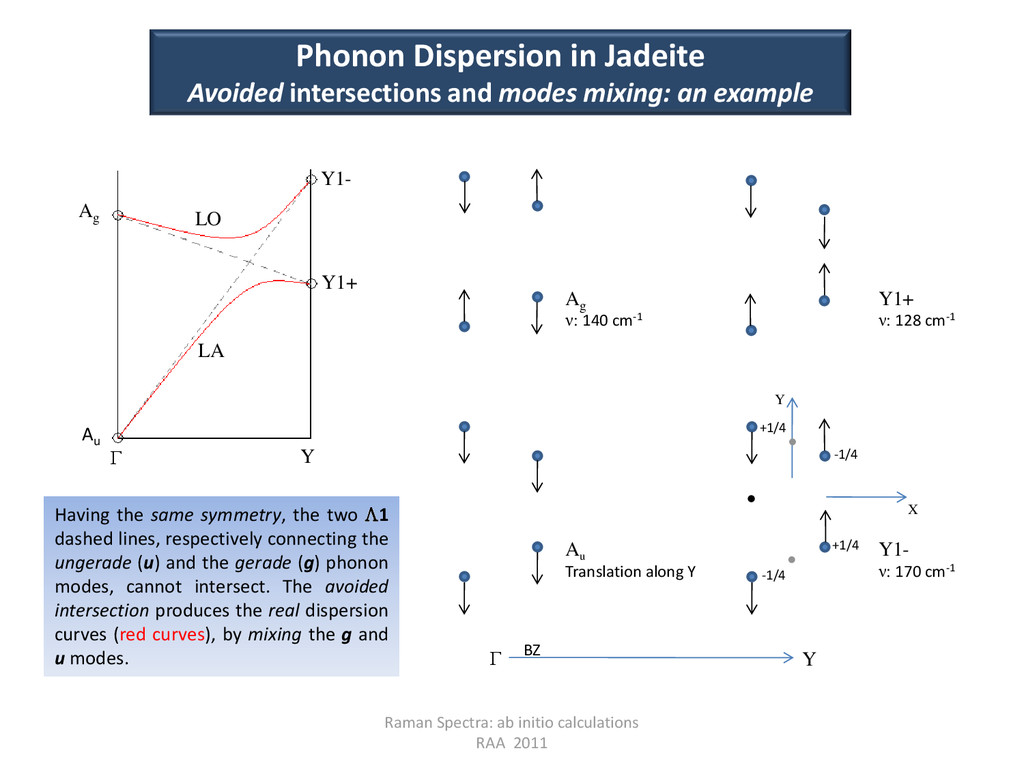
Y1+ Y1- LA LO Г Phonon Dispersion in Jadeite Avoided intersections and modes mixing: an example Having the same symmetry, the two 1 dashed lines, respectively connecting the ungerade (u) and the gerade (g) phonon modes, cannot intersect. The avoided intersection produces the real dispersion curves (red curves), by mixing the g and u modes. +1/4 +1/4 -1/4 -1/4 X Y Au Translation along Y Y1- ν: 170 cm-1 Ag ν: 140 cm-1 Y1+ ν: 128 cm-1 Г Y BZ
G and Y Modes: the G/Y-Modes Overlap N i i B 3 , 1 ) ( G G q N i i B 3 , 1 ) ( q and G Y a b Vectors from the EG and EY 3N-D subspaces (where N is the number of atoms in the unit cell), respectively spanned by the G and Y-orthogonal eigenvectors, can be projected onto the Ea subspace defined by the N 3D cartesian frames centered on each atom in the unit cell (a). The two sets are not reciprocally orthogonal, and they are both orthogonal bases of the 3N-D Ea subspace. ) ( ), ( G G G E E i j j j ij i q q q q Each vector of the projected EY space can be expressed as a linear combination of the vectors BG. In particular: The coefficients can be used to relate G and Y eigenvectors, as they express the overlap between the G and Y modes a a : : E E E E G
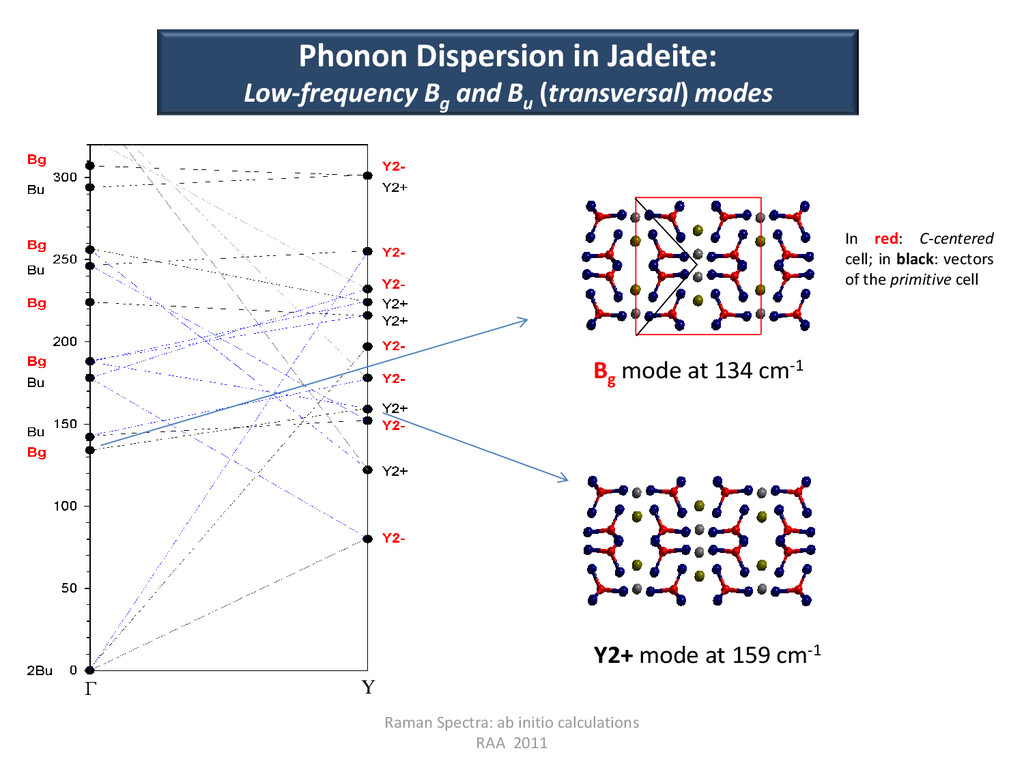
Raman Spectra: ab initio calculations RAA 2011 Bg mode at 134 cm-1 Y2+ mode at 159 cm-1 Γ Y In red: C-centered cell; in black: vectors of the primitive cell
-1/4 -1/4 -1/4 -1/4 +1/4 +1/4 +1/4 +1/4 -1/4 n 2 2 +1/4 +1/4 X Y Omphacite: structure and relations to jadeite Ordered omphacite: (NaCa)(AlMg)Si4 O12 Space group P2/n One half of the Na atoms in jadeite are substituted by Ca atoms, whereas one half of the Al atoms are substituted by Mg atoms; the ordering of the cations in the structural sites of omphacite removes the C centering of the cell and some of the inversion centers; by convention, the origin of the cell is shifted on the inversion center; metrically, the primitive cell of the ordered omphacite can be seen as based on the C centered cell of jadeite, with the removal of an inversion center (and the shift and the transformation of the c glide in a n glide) Trace of the C-centered cell of jadeite
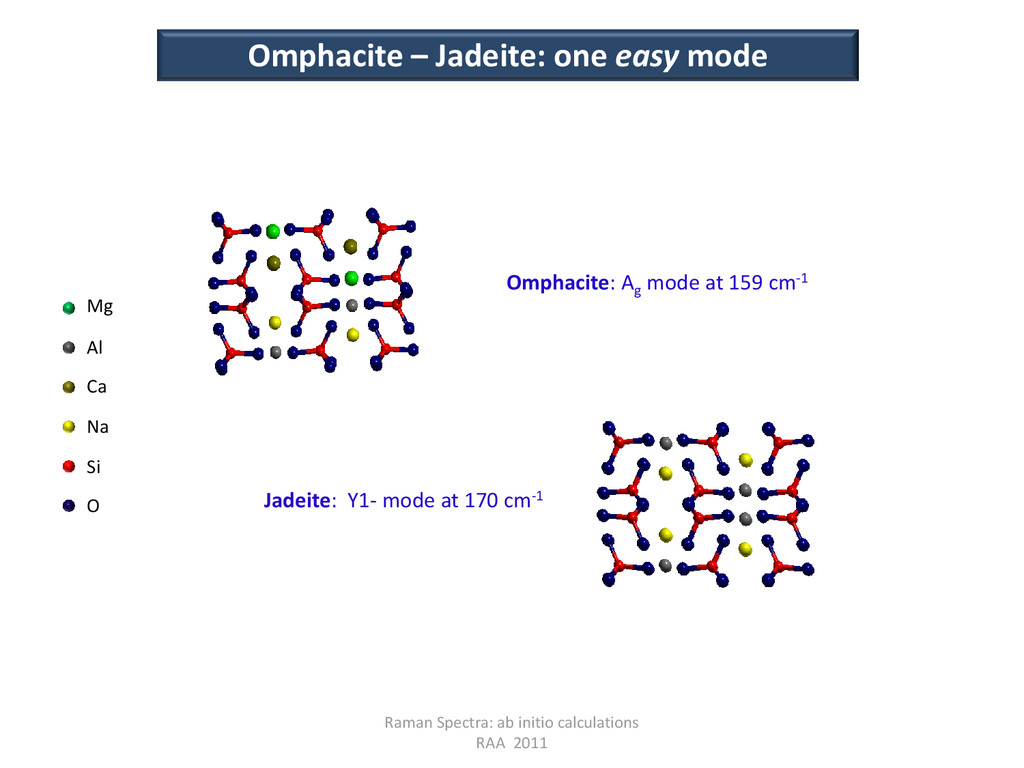
Y1- mode in jadeite Raman Spectra: ab initio calculations RAA 2011 Omphacite: symmetry of the modes Jadeite The modes at the G point, are respectively symmetric or anti-symmetric with respect to all of the inversion centers in the structure; at the Y point, due to the modulation wave, atoms at x=1/2 are no longer equivalent to the atoms at x=0, and the inversion center at x=1/4 is lost. Omphacite The inversion center at x=1/4 of jadeite, becomes the unique inversion center of omphacite, and it is shifted on the origin of the cell; it appears that G-modes in jadeite have the same parity of the corresponding modes in omphacite; whereas Y-modes in jadeite have opposite parity with respect to the corresponding G-modes in omphacite. Jadeite → Omphacite Ag Ag Au Au Bg Bg Bu Bu Y1+ Au Y1- Ag Y2+ Bu Y2- Bg
modes Jadeite Omphacite Diopside Notes Sym ν (cm-1) Sym ν Sym ν Au 0 Au 0 Au 0 Translation along Y Y1+ 129 Au 131 Y1+ 130 LA Y-mode in Jd and Di Ag 141 Ag 143 Ag 141 LO mode Y1- 170 Ag 159 Y1- 152 LO Y-mode in Jd and Di Longitudinal modes Frequencies of diopside (C2/c; CaMgSi2 O6 ) are also reported. The new Raman active Ag mode at 159 cm-1, in omphacite, appears to be relatively strongly dependent by the Ca content (and perhaps by ordering), at variance with the other lower frequency modes. Jadeite Omphacite Diopside Notes Sym ν (cm-1) Sym ν Sym ν 2Bu 0 2Bu 0 2Bu 0 Translations along X, Z Y2- 81 Bg 89 Y2- 99 TA Y-mode in Jd and Di Y2+ 122 Bu 123 Y2+ 123 TA Y-mode in Jd and Di Transversal modes In omphacite, a new Raman active Bg mode appears at 89 cm-1.
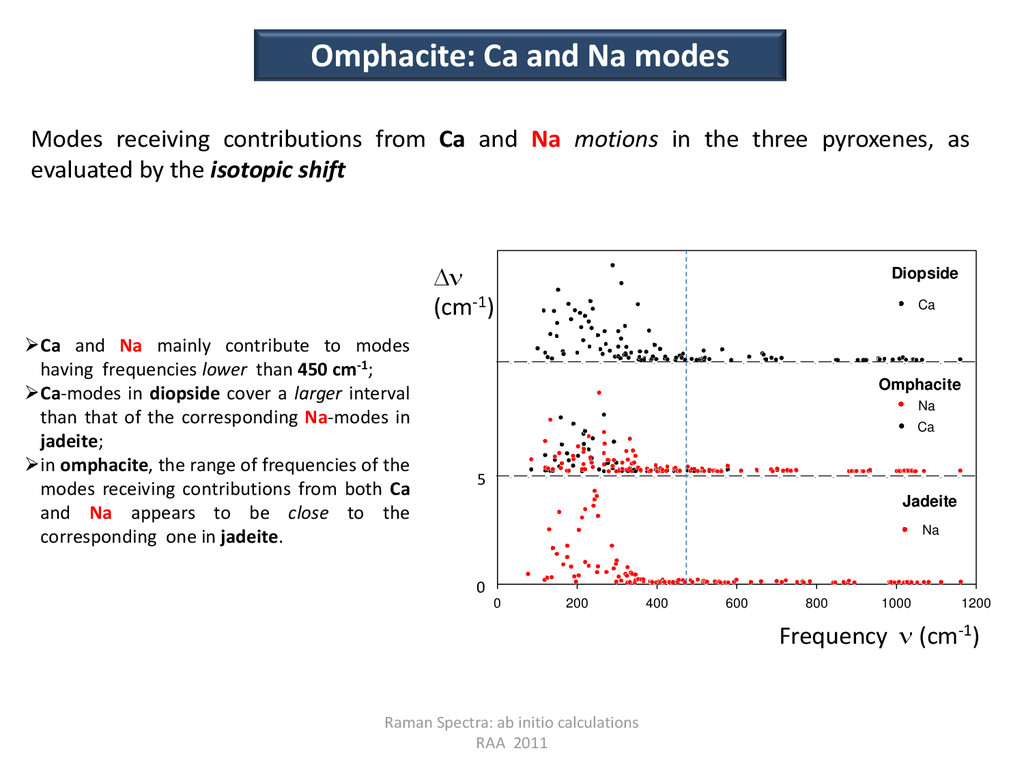
Na modes Modes receiving contributions from Ca and Na motions in the three pyroxenes, as evaluated by the isotopic shift 0 200 400 600 800 1000 1200 Jadeite Omphacite Diopside Na Na Ca Ca Frequency (cm-1) D (cm-1) 0 5 Ca and Na mainly contribute to modes having frequencies lower than 450 cm-1; Ca-modes in diopside cover a larger interval than that of the corresponding Na-modes in jadeite; in omphacite, the range of frequencies of the modes receiving contributions from both Ca and Na appears to be close to the corresponding one in jadeite.
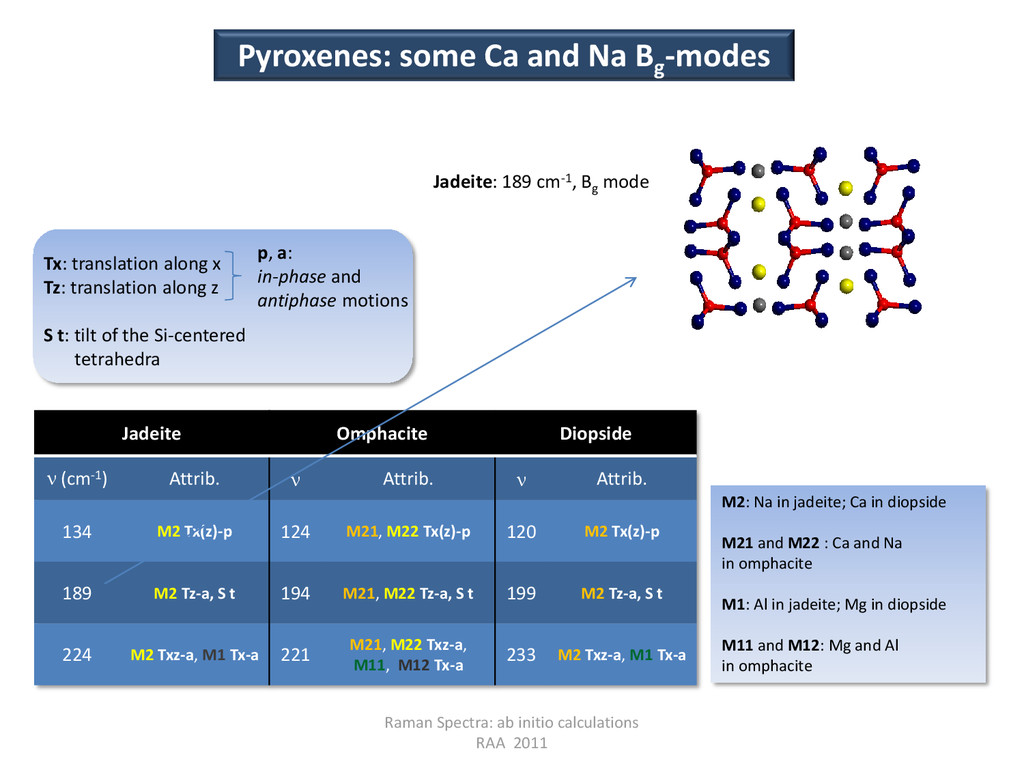
(cm-1) Attrib. Attrib. Attrib. 134 M2 Tx(z)-p 124 M21, M22 Tx(z)-p 120 M2 Tx(z)-p 189 M2 Tz-a, S t 194 M21, M22 Tz-a, S t 199 M2 Tz-a, S t 224 M2 Txz-a, M1 Tx-a 221 M21, M22 Txz-a, M11, M12 Tx-a 233 M2 Txz-a, M1 Tx-a Pyroxenes: some Ca and Na Bg -modes Jadeite: 189 cm-1, Bg mode M2: Na in jadeite; Ca in diopside M21 and M22 : Ca and Na in omphacite M1: Al in jadeite; Mg in diopside M11 and M12: Mg and Al in omphacite Tx: translation along x Tz: translation along z S t: tilt of the Si-centered tetrahedra p, a: in-phase and antiphase motions
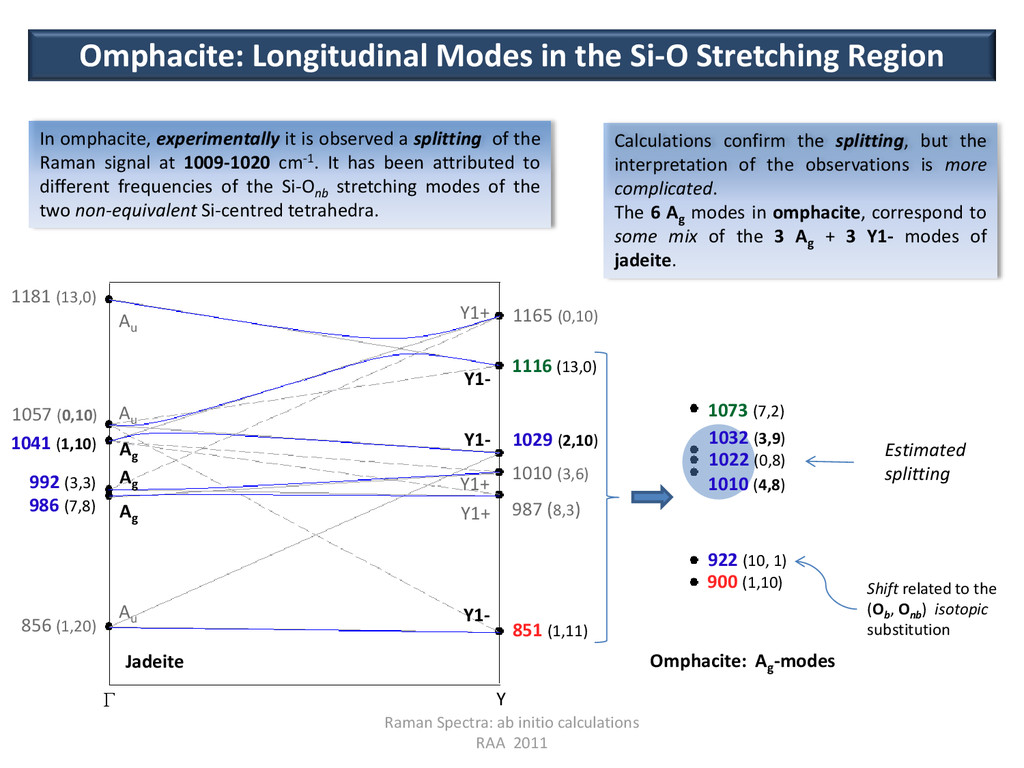
Ag Ag Ag Y1- Y1- Y1- Y1+ Y1+ Y1+ 851 (1,11) 987 (8,3) 1010 (3,6) 1029 (2,10) 1116 (13,0) 1165 (0,10) 856 (1,20) 992 (3,3) 986 (7,8) 1041 (1,10) 1057 (0,10) 1181 (13,0) Jadeite 900 (1,10) 922 (10, 1) 1073 (7,2) Omphacite: Ag -modes Estimated splitting Shift related to the (Ob , Onb ) isotopic substitution 1032 (3,9) 1022 (0,8) 1010 (4,8) G Y Omphacite: Longitudinal Modes in the Si-O Stretching Region In omphacite, experimentally it is observed a splitting of the Raman signal at 1009-1020 cm-1. It has been attributed to different frequencies of the Si-Onb stretching modes of the two non-equivalent Si-centred tetrahedra. Calculations confirm the splitting, but the interpretation of the observations is more complicated. The 6 Ag modes in omphacite, correspond to some mix of the 3 Ag + 3 Y1- modes of jadeite.
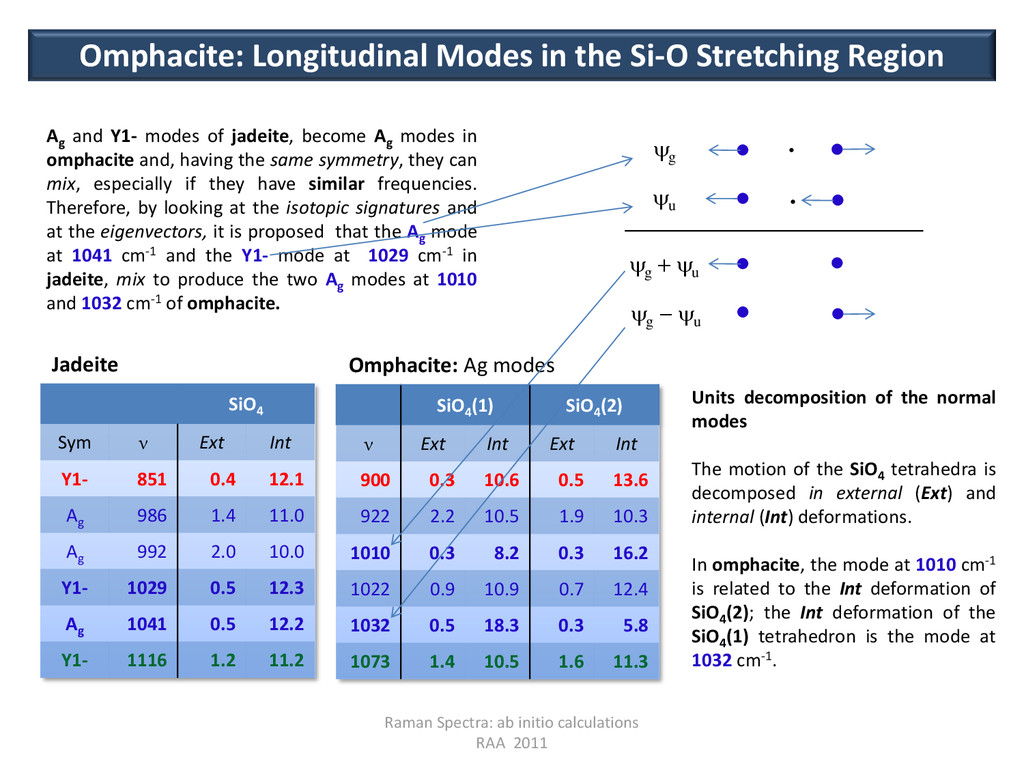
(2) ν Ext Int Ext Int 900 0.3 10.6 0.5 13.6 922 2.2 10.5 1.9 10.3 1010 0.3 8.2 0.3 16.2 1022 0.9 10.9 0.7 12.4 1032 0.5 18.3 0.3 5.8 1073 1.4 10.5 1.6 11.3 Omphacite: Ag modes SiO4 Sym ν Ext Int Y1- 851 0.4 12.1 Ag 986 1.4 11.0 Ag 992 2.0 10.0 Y1- 1029 0.5 12.3 Ag 1041 0.5 12.2 Y1- 1116 1.2 11.2 Omphacite: Longitudinal Modes in the Si-O Stretching Region Jadeite Ag and Y1- modes of jadeite, become Ag modes in omphacite and, having the same symmetry, they can mix, especially if they have similar frequencies. Therefore, by looking at the isotopic signatures and at the eigenvectors, it is proposed that the Ag mode at 1041 cm-1 and the Y1- mode at 1029 cm-1 in jadeite, mix to produce the two Ag modes at 1010 and 1032 cm-1 of omphacite. g u g u g u Units decomposition of the normal modes The motion of the SiO4 tetrahedra is decomposed in external (Ext) and internal (Int) deformations. In omphacite, the mode at 1010 cm-1 is related to the Int deformation of SiO4 (2); the Int deformation of the SiO4 (1) tetrahedron is the mode at 1032 cm-1.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}