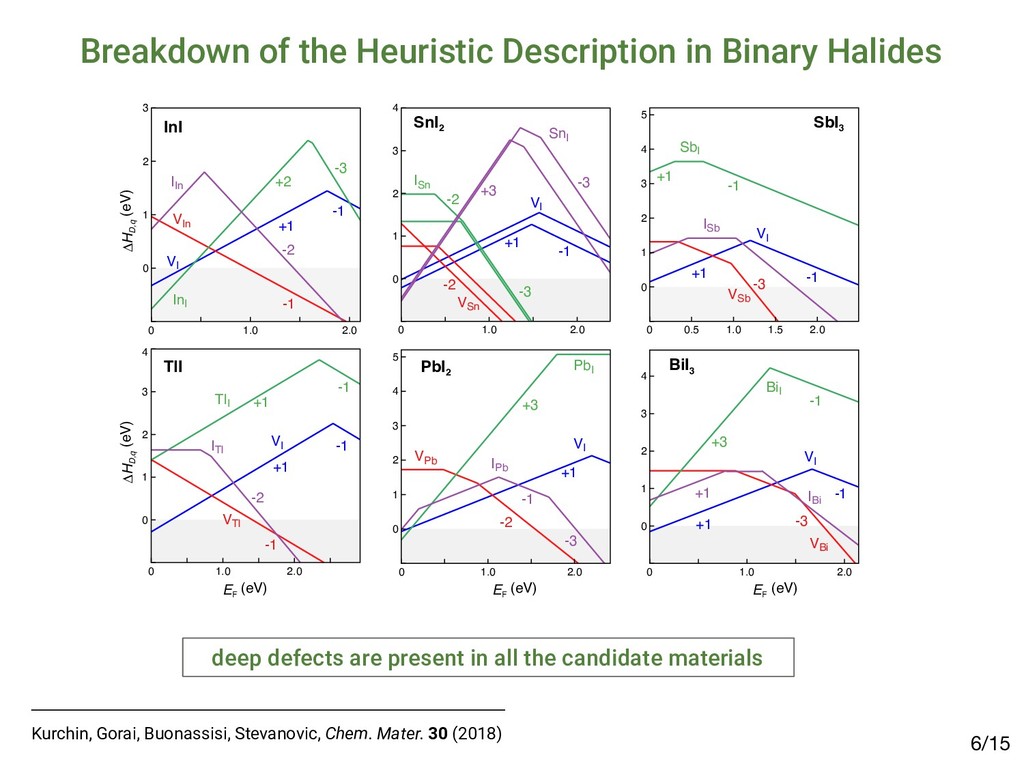
on heuristic guidelines identified several candidates1: • Binary halides: InI, TlI, PbI2, SnI2, SbI3, BiI3 • Chalcohalides: BiOI, BiSI, BiSeI, SbSI, SbSeI, … Prospective Article Identifying defect-tolerant semiconductors with high minority-carrier lifetimes: beyond hybrid lead halide perovskites Riley E. Brandt, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA Vladan Stevanović, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, Colorado 80401; Colorado School of Mines, 1500 Illinois Street, Golden, Colorado 80401, USA David S. Ginley, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, Colorado 80401, USA Tonio Buonassisi, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA Address all correspondence to Riley E. Brandt, Tonio Buonassisi at
[email protected];
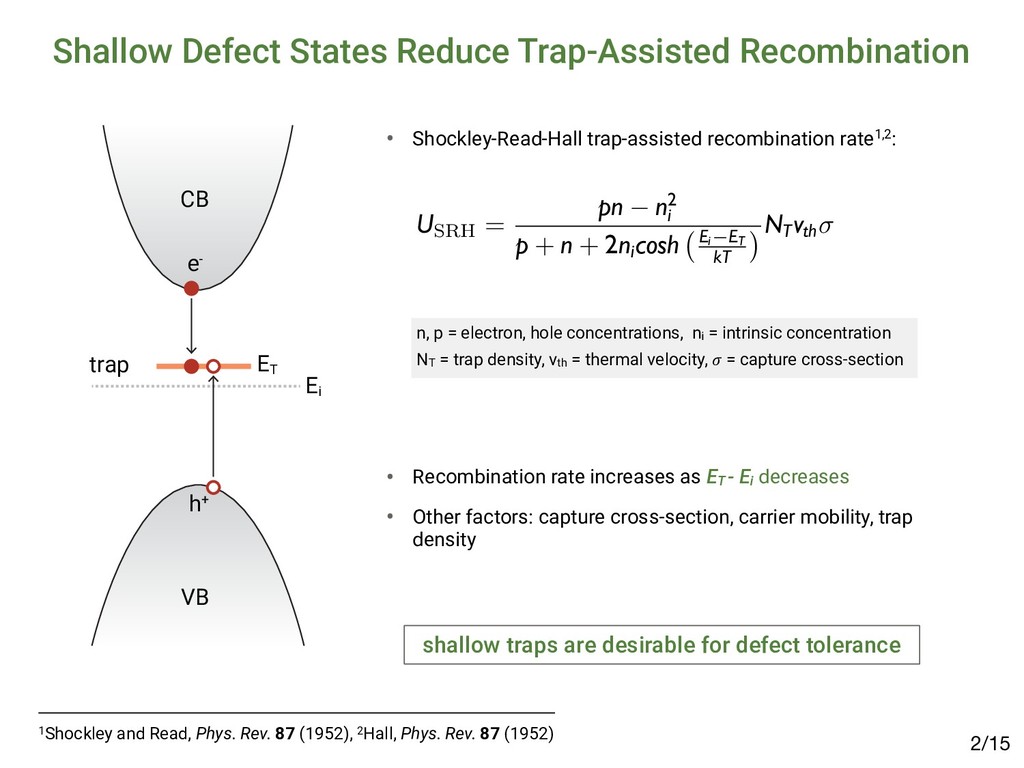
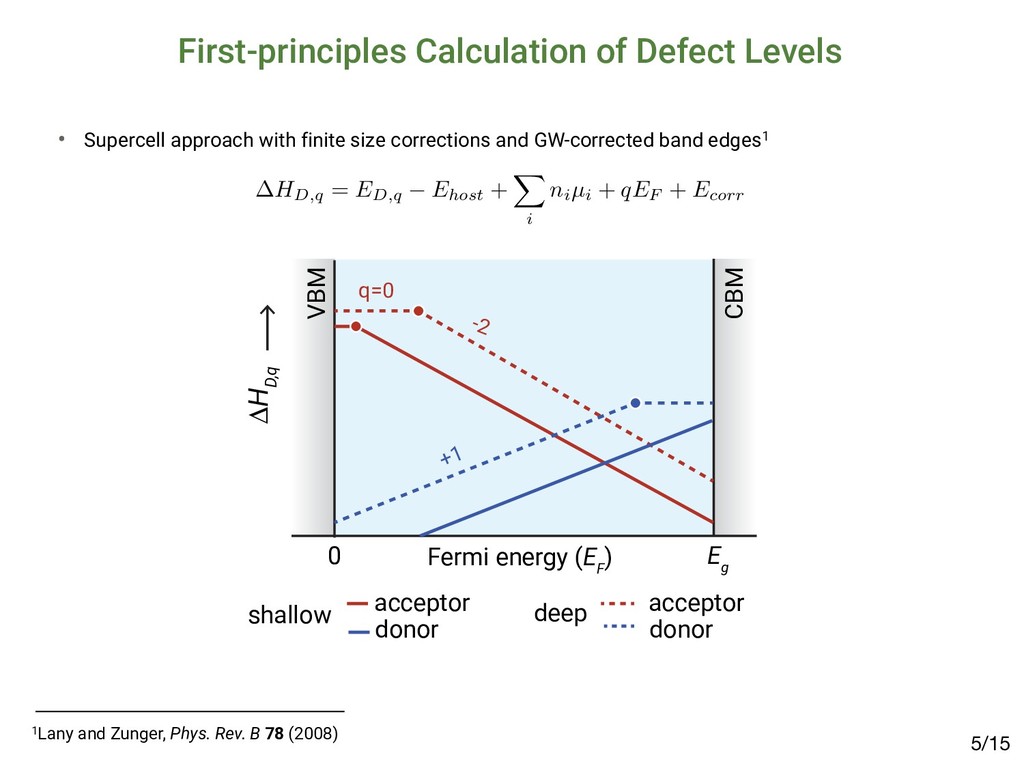
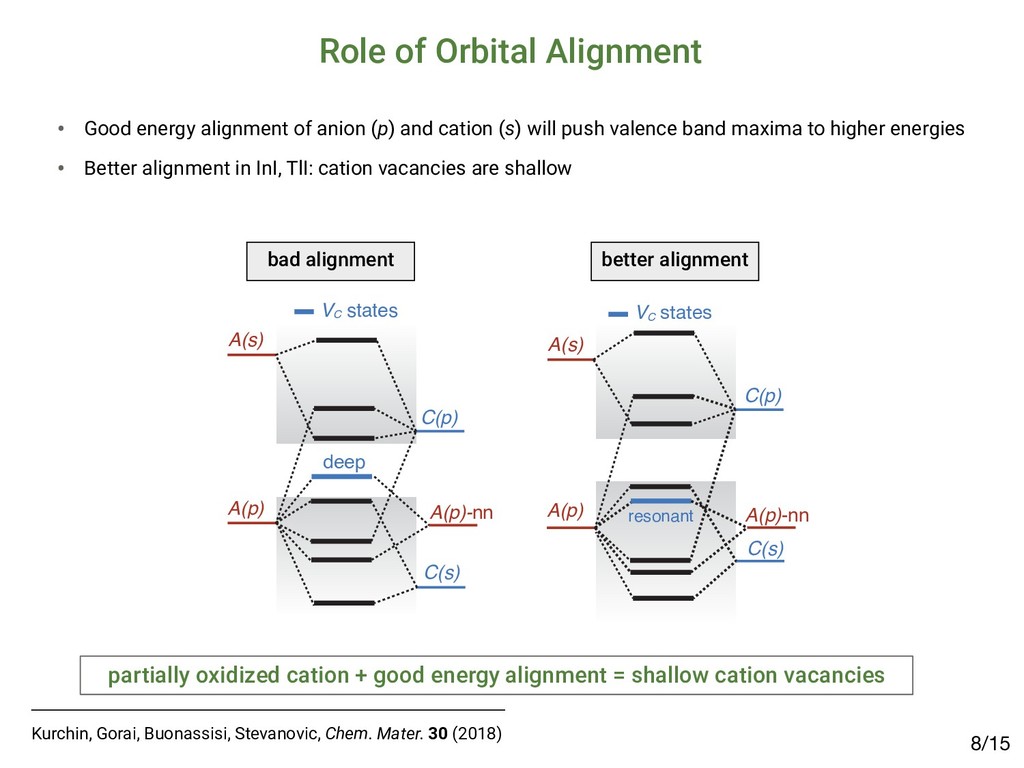
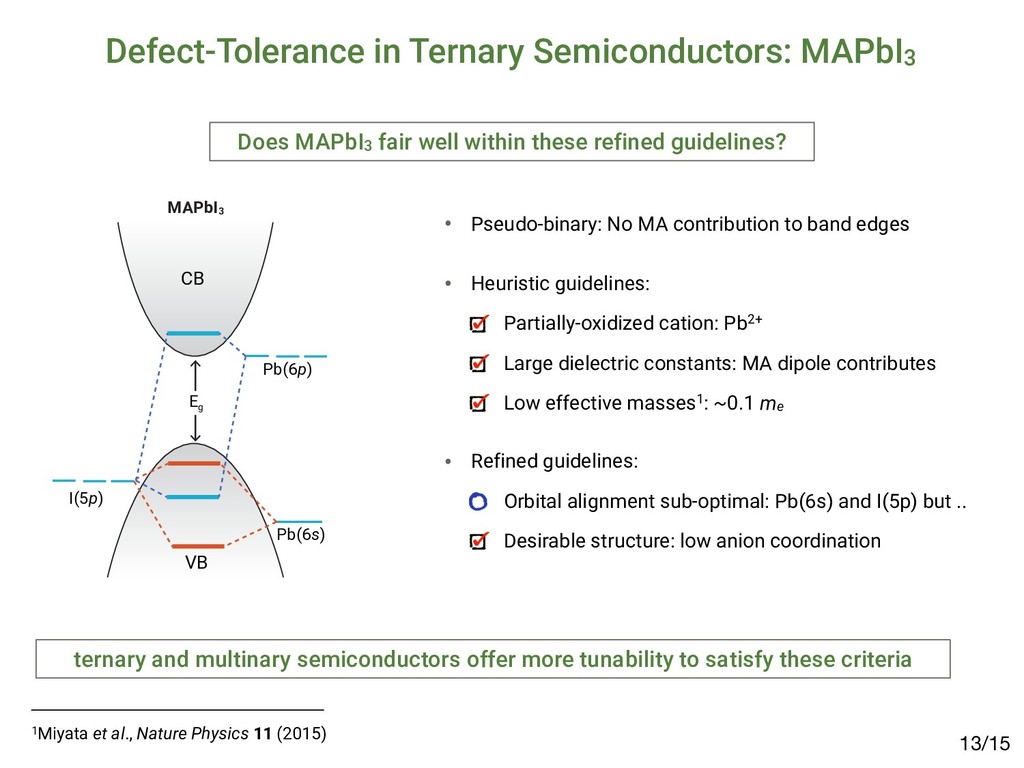
[email protected] (Received 27 March 2015; accepted 23 April 2015) Abstract The emergence of methyl-ammonium lead halide (MAPbX3 ) perovskites motivates the identification of unique properties giving rise to excep- tional bulk transport properties, and identifying future materials with similar properties. Here, we propose that this “defect tolerance” emerges from fundamental electronic-structure properties, including the orbital character of the conduction and valence band extrema, the charge- carrier effective masses, and the static dielectric constant. We use MaterialsProject.org searches and detailed electronic-structure calculations to demonstrate these properties in other materials than MAPbX3 . This framework of materials discovery may be applied more broadly, to accelerate discovery of new semiconductors based on emerging understanding of recent successes. Introduction Many semiconductors have been studied over the last century for their possible use in photovoltaics (PVs), light-emitting diodes (LEDs), computing devices, sensors, and detectors. Of these, only a select few have achieved sufficient optoelectronic perfor- mance to transition into industrial manufacturing, and their iden- tification and development have been slow and mostly empirical. Recently, high-throughput computation and a deeper under- standing of the physics-based requirements for high performance have created the potential for an accelerated identification of functional materials with manufacturing potential. For example, materials screening criteria have been proposed to better focus the search for novel candidate PV materials; they include the op- tical band-gap energy[1,2] and abruptness of absorption onset,[3,4] in addition to boundary conditions of elemental abundance,[5] and manufacturing cost.[6,7] However, many materials have met these criteria and yet have not achieved industrially relevant conversion efficiencies (in excess of 10–15%) due to low minority-carrier lifetimes or diffusion lengths, e.g., in the case of Cu2 ZnSn(S,Se)4 ,[8] SnS,[9] and others.[10] Meanwhile, PV de- vices have emerged based on methyl-ammonium lead iodide (MAPbI3 ) and closely related halides (herein referred to as MAPbX3 ). MAPbI3 is a semiconductor, which has demonstrated exceptional minority-carrier lifetimes of 280 ns (in the mixed iodide–chloride composition)[11] and diffusion lengths up to 175 μm,[12] comparable with the best single-crystal semiconduc- tors. This, in addition to meeting the criteria above, has resulted in a dramatic realization of PV conversion efficiencies up to 20.1%[13,14] in 2015, from <4% in 2009.[15] This paper examines whether the dramatic success of MAPbX3 in PVs can be used as a basis to expand design criteria to identify new potential high-performance optoelectronic ma- terials. One of the most compelling questions engendered by MAPbX3 as an optoelectronic material is the degree to which it is unique, and whether its success can lead to the identifica- tion of materials with improved stability and lower toxicity, yet similar high performance. Clearly, as in previous design criteria the high optical absorption coefficient is important, but also es- sential are the long carrier diffusion lengths observed in MAPbX3 , enabled by high minority-carrier lifetime (τ) and mo- bility (μ).[11,16,17] The importance of τ and μ for device perfor- mance has been established[18] for the most highly performing PV materials, including silicon,[19] cadmium telluride,[20,21] copper indium gallium diselenide,[22] and gallium arsenide.[23] Oddly, these more direct transport parameters, τ and μ, are not traditionally considered essential screening criteria for novel candidate PV materials. This may be partially a conse- quence of the difficulty in measuring and/or calculating these parameters. The direct measurement of minority carrier τ and μ requires ultrafast electronic or optical sensors to capture tran- sients,[24–27] or strong steady-state signals,[28,29] and must be performed with PV-device-relevant illumination conditions, electrical fields, and transport directions. Calculating τ and μ from first-principles is even more challenging, given the lack of well-established and high-throughput methods to directly calculate electron–phonon interactions and/or trap capture cross-sections. Although it is possible to obtain some informa- tion about carrier mobility from effective masses, and MRS Communications (2015), 5, 265–275 doi:10.1557/mrc.2015.26 MRS COMMUNICATIONS • VOLUME 5 • ISSUE 2 • www.mrs.org/mrc ▪265 1Brandt et al., MRS Comm. 5 (2015); 2Brandt et al., Chem. Mater. 29 (2017) Searching for “Defect-Tolerant” Photovoltaic Materials: Combined Theoretical and Experimental Screening Riley E. Brandt,*,† Jeremy R. Poindexter,† Prashun Gorai,‡,§ Rachel C. Kurchin,† Robert L. Z. Hoye,†,@ Lea Nienhaus,† Mark W. B. Wilson,†,# J. Alexander Polizzotti,† Raimundas Sereika,∥ Raimundas Žaltauskas,∥ Lana C. Lee,⊥ Judith L. MacManus-Driscoll,⊥ Moungi Bawendi,† Vladan Stevanović,‡,§ and Tonio Buonassisi† † Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States ‡ Colorado School of Mines, Golden, Colorado 80401, United States §National Renewable Energy Laboratory, Golden, Colorado 80401, United States ∥Faculty of Science and Technology, Lithuanian University of Educational Sciences, Vilnius 08106, Lithuania ⊥Department of Materials Science and Metallurgy, University of Cambridge, Cambridge CB3 0FE, United Kingdom * S Supporting Information ABSTRACT: Recently, we and others have proposed screening criteria for “defect-tolerant” photovoltaic (PV) absorbers, identifying several classes of semiconducting compounds with electronic structures similar to those of hybrid lead−halide perovskites. In this work, we reflect on the accuracy and prospects of these new design criteria through a combined experimental and theoretical approach. We construct a model to extract photoluminescence lifetimes of six of these candidate PV absorbers, including four (InI, SbSI, SbSeI, and BiOI) for which time-resolved photoluminescence has not been previously reported. The lifetimes of all six candidate materials exceed 1 ns, a threshold for promising early stage PV device performance. However, there are variations between these materials, and none achieve lifetimes as high as those of the hybrid lead−halide perovskites, suggesting that the heuristics for defect-tolerant semiconductors are incomplete. We explore this through first- principles point defect calculations and Shockley−Read−Hall recombination models to describe the variation between the measured materials. In light of these insights, we discuss the evolution of screening criteria for defect tolerance and high-performance PV materials. ▪ INTRODUCTION Thin-film polycrystalline photovoltaic (PV) materials offer the potential for lower-capital intensity manufacturing relative to crystalline silicon PV, but only if they can achieve high PV conversion efficiencies in excess of 20%.1,2 Long minority- carrier lifetimes are necessary to achieve high efficiency, yet to date, there have been only a few classes of polycrystalline semiconductors that have demonstrated minority-carrier life- times in excess of 1 ns.3 Three classes of thin-film PV absorbers have achieved bulk lifetimes in excess of 100 ns, and they materials: it allows power conversion efficiencies on the order of 10% in materials with strong optical absorption and suggests the potential for further improvements.3 Including InP and other III−V materials, as well as the Cu2 ZnSn(S,Se)4 family of materials14 in addition to CIGS, CdTe, and LHPs, there are several additional classes of thin-film inorganic (or hybrid organic−inorganic) polycrystalline semiconductors that are known to exceed this 1 ns threshold. In prior work, we hypothesized that a promising path to achieving these long lifetimes is through defect-tolerant Article pubs.acs.org/cm • Candidates exhibit lifetimes >1 nanosecond2 but not comparable to MAPbI3: ▪ RESULTS Photoluminescence Lifetime. TRPL data for all six materials are plotted in Figure 1. There is a clear variation in the TRPL behavior across the materials tested, with (CH NH ) Bi I and InI demonstrating the slowest decay, however, all effective lifetimes as well as “slow” expo time constants exceed 1 ns. Defect Calculations. Figure 2 shows calculated formation enthalpies as a function of Fermi level for I Figure 1. TRPL decay curves of all six materials measured. Dashed gray lines represent biexponential fits to the data, while solid black lines using a numerical model incorporating both Shockley−Read−Hall and radiative recombination (see the Supporting Information for det estimated parameters). The fluence used was 20 nJ/cm2 for all traces except for those of (CH3 NH3 )3 Bi2 I9 (approximately 1 nJ/cm2) and S nJ/cm2). Data for (CH3 NH3 )3 Bi2 I9 were reproduced with permission from ref 33. Chemistry of Materials A defect calculations can reveal the nature of defects /15 4
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}