and complexities ▸ Every genome assembly and annotation is truly de novo: no reference genomes ▸ We often need special techniques: hybrid assembly, lots of long-read sequencing to get a contiguous genome worth annotating ▸ Usually less funding out there on the lonely branches of the tree of life ▸ Not everyone has HPC
Science Team (Office of the CIO) Beth & Dan: OCIO Office of Research Information Services Deron: CIO Intel Corporation Sandeep: Platform Applications Engineer Mathew: Senior Solutions Strategist and Architect Amazon Web Services Stephanie: Federal Account Manager Dave: Senior Solution Architect Dikow, R. B., Gupta, S., Taylor, M. H. 2016. Accelerating Plant and Animal Genomics for Biodiversity with the Latest Intel Technologies. Intel Corporation white paper.
▸ Annotate 25 Smithsonian genomes in 12 months ▸ Figure out the best way to do implement existing annotation pipelines in the cloud ▸ Write our own pipeline that takes advantage of cloud strengths ▸ fast AND easy to spin-up
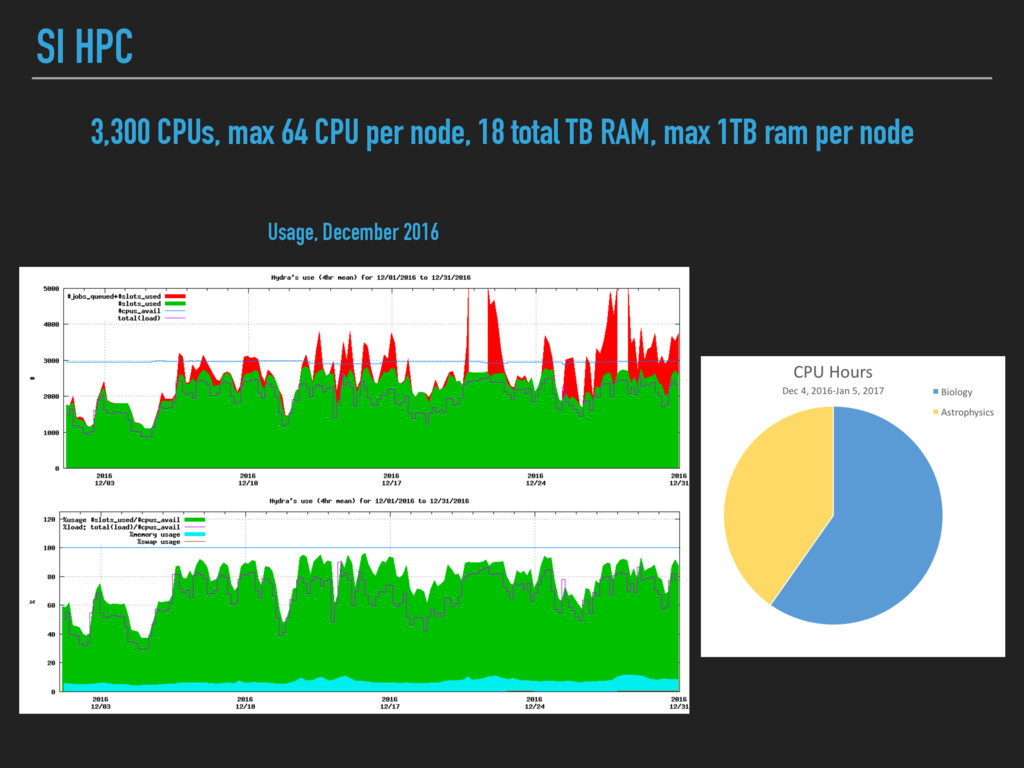
- but you have to be careful! ▸ Can easily deploy other people’s AMIs, use Docker containers ▸ HPC often appears free to most users (but it’s not!) ▸ Often not agile in terms of software installation ▸ Queue limits ▸ When it’s full, it’s full
check out HC Lim’s talk! check out Mirian Tsuchiya’s poster! Other Smithsonian genomes: Amakihi Golden collared manakin Raccoon Heliconius spp. Greater bamboo lemur …
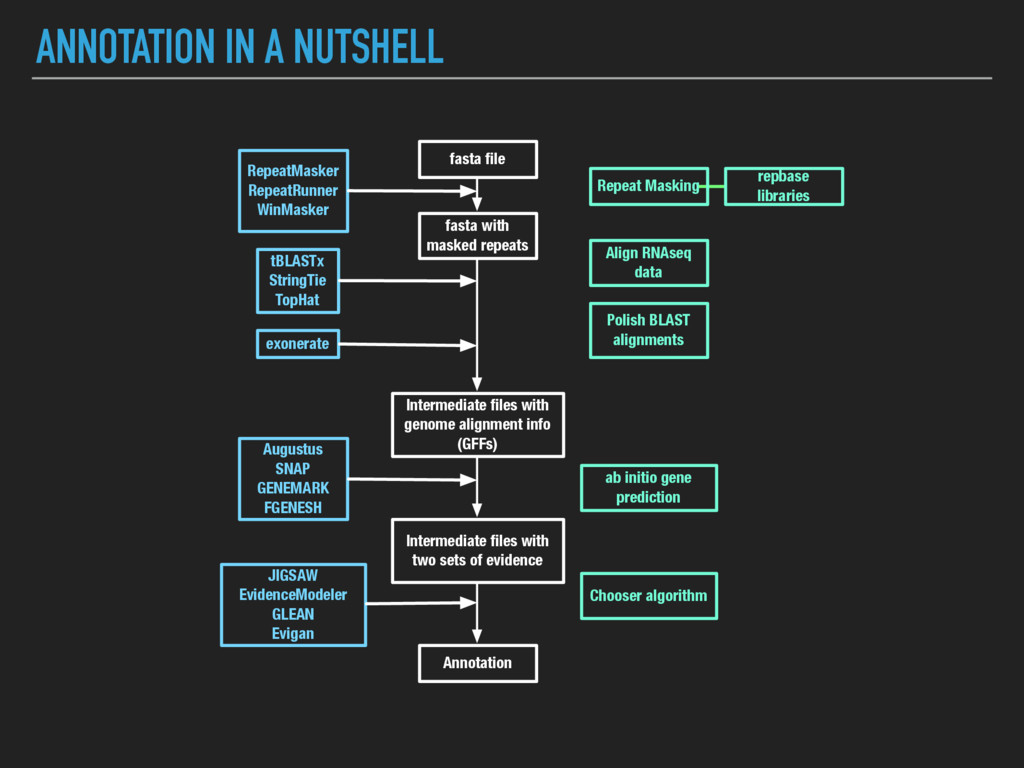
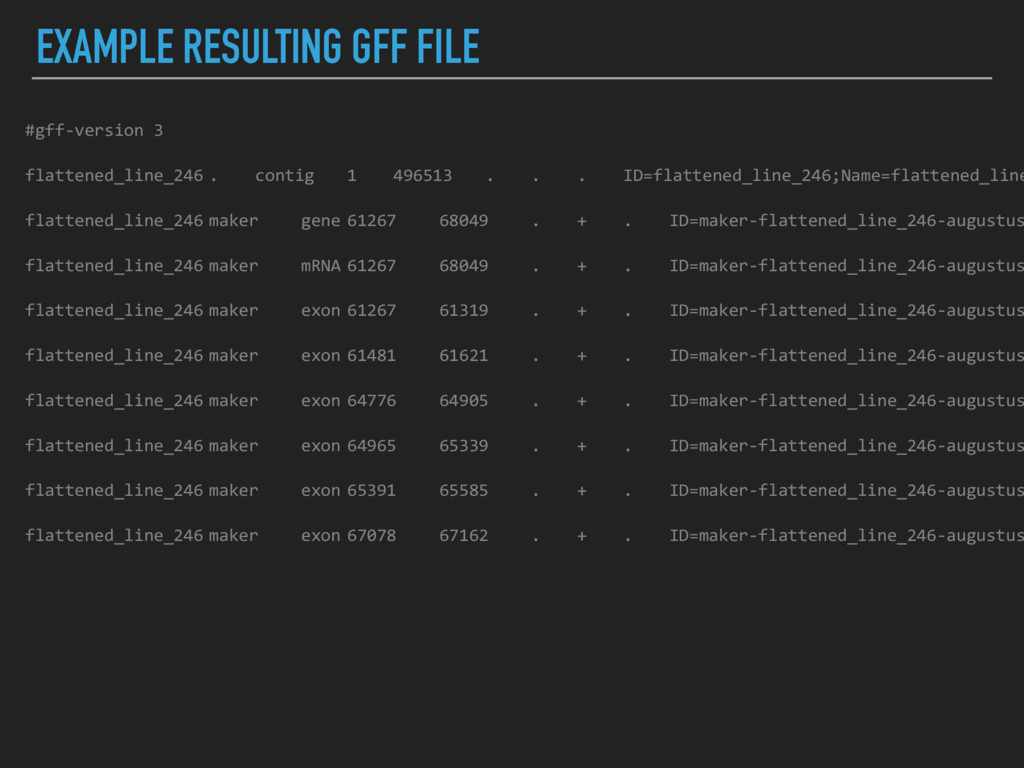
2011) came out in 2008 ▸ Major steps: identifies repeats, aligns ESTs and proteins to a genome, produces ab initio gene predictions, and synthesizes these into gene annotations ▸ Operationally, each of these processes is performed on each contig in turn, producing a GFF file (General Feature Format) for each contig ▸ Large contigs are “chunked” into smaller pieces and then the GFFs are knitted together to optimize compute time (enable more operations to run in parallel) and to save RAM
parallelization (each contig can in theory be processed simultaneously), but implementation is a bit tricky ▸ MAKER2 allows parallelization using MPI, but has problems utilizing NFS (Network File System) data storage, making it hard to run on HPC. ▸ It’s also frustrating to install, with lots of dependencies: ▸ BioPerl ▸ RepeatMasker ▸ exonerate ▸ SNAP ▸ Augustus ▸ OpenMPI
master-worker applications that span thousands of machines drawn from clusters, clouds, and grids. ▸ Thrasher et al., 2012 implements MAKER in the Work Queue framework (wq-maker).
install Work Queue, MAKER, and dependencies ▸ Save your environment as an AMI (Amazon Machine Image) ▸ Spin up as many workers with that same AMI as you’d like ▸ Run wq-maker on the master node and send off jobs to the workers ▸ Results are written to the master node ▸ While we were working on this, we saw a presentation by the CyVerse folks showing a similar implementation in Jetstream (XSEDE’s “cloud”) and tested that too after requesting an allocation.
Playbook ▸ Ansible is an open source automation platform ▸ Under active development and the developers are very friendly! ▸ https://wiki.cyverse.org/wiki/display/TUT/MAKER+2.31.8+with+CCTOOLS+Jetstream +Tutorial
to “instance store” or “ephemeral” storage ▸ Augustus config directory with gene models must be in the working directory ▸ wq-maker relies on MAKER 2.31.8 ▸ The Augustus version that you can install through MAKER is not the most recent version - you should install separately to get all the gene models
to keep AMIs and/or Jetstream instances updated ▸ Dependencies are complex and very specific ▸ Pipeline is not modular - it runs from start to finish ▸ MAKER was written for a specific use case and has been built out to accomplish more with legacy code
written in Python, uses Common Workflow Language definitions ▸ First Toil paper: analyzed 20,000 RNAseq samples on 32,000 cores in 4 days (Vivian et al. 2016 bioRxiv doi: 10.1101/062497) ‣ Why we like Toil: ▸ can also be used on HPC, local workstation, or laptop ▸ fault tolerant ▸ easy to install - can provide virtual machines or containerize ▸ steps are modular steps because it uses CWL
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}