information from (large) genomic datasets • Embarrassingly parallelisation, can spawn 100s-100k jobs over distributed cluster • Mash-up of many different tools and scripts (dependancies!) • Complex dependency trees and configuration → very fragile ecosystem
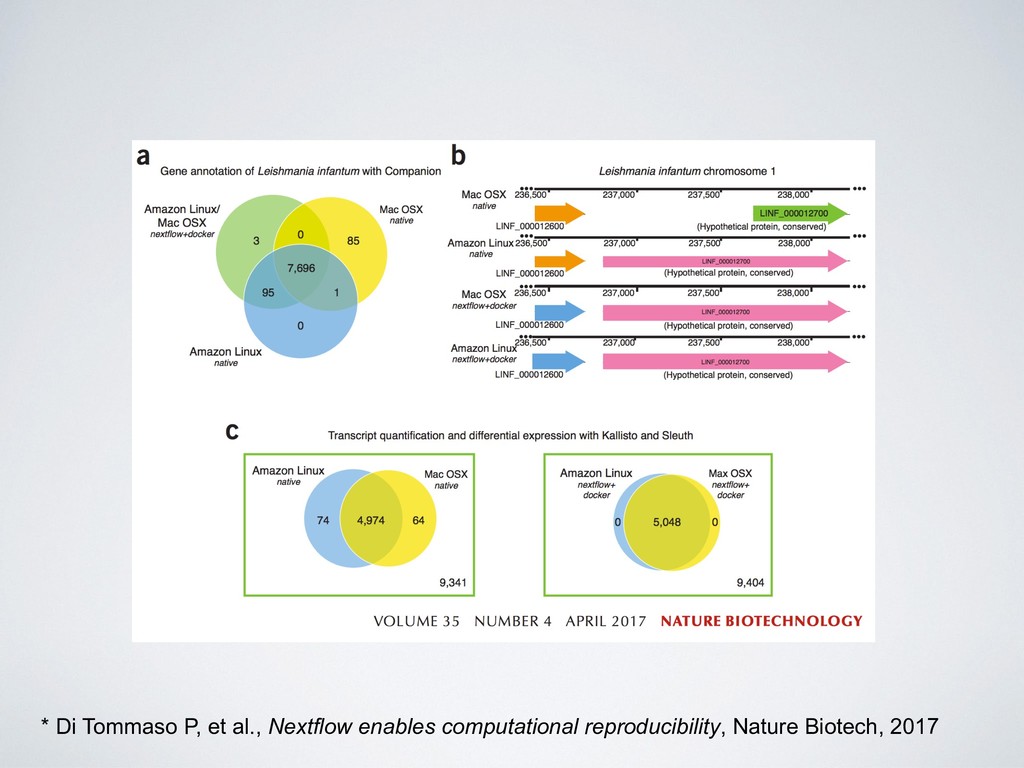
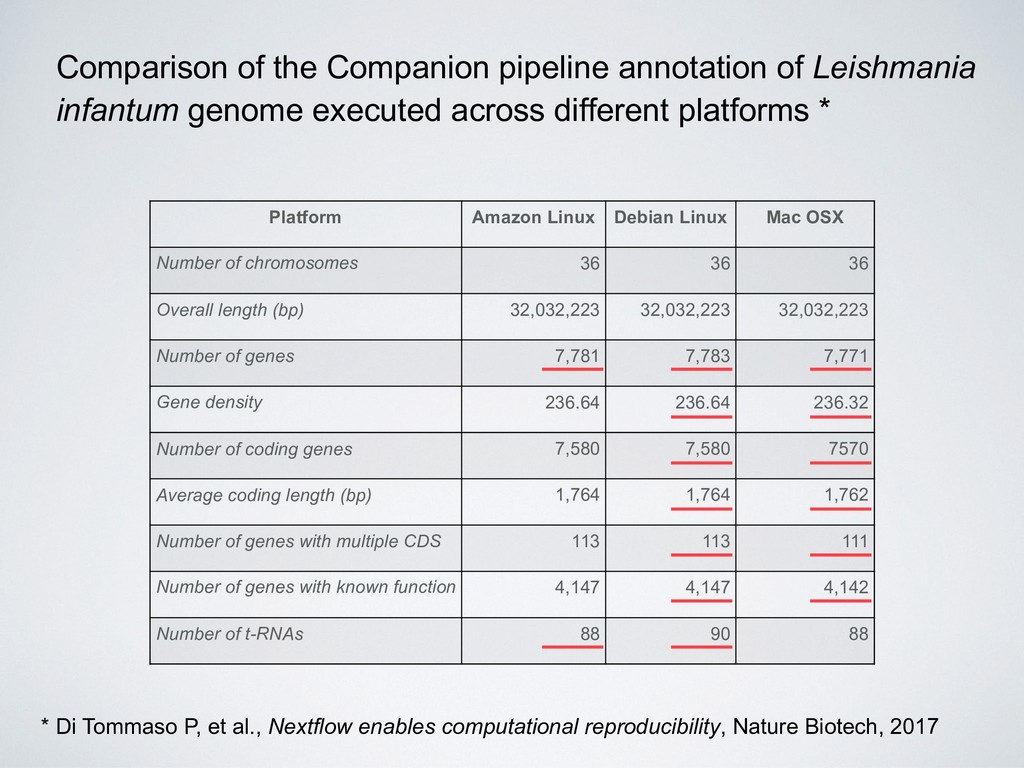
36 36 36 Overall length (bp) 32,032,223 32,032,223 32,032,223 Number of genes 7,781 7,783 7,771 Gene density 236.64 236.64 236.32 Number of coding genes 7,580 7,580 7570 Average coding length (bp) 1,764 1,764 1,762 Number of genes with multiple CDS 113 113 111 Number of genes with known function 4,147 4,147 4,142 Number of t-RNAs 88 90 88 Comparison of the Companion pipeline annotation of Leishmania infantum genome executed across different platforms * * Di Tommaso P, et al., Nextflow enables computational reproducibility, Nature Biotech, 2017
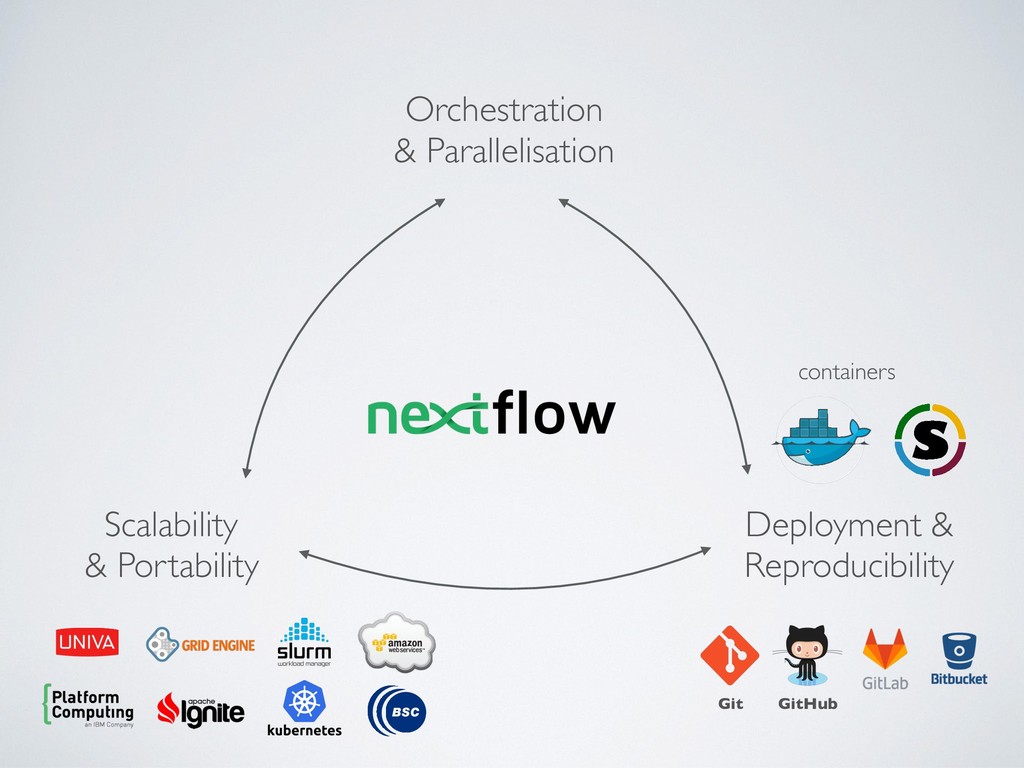
across different platforms • Scalability ie. deploy big distributed workloads • Usability, streamline execution and deployment of complex workloads ie. remove complexity instead of adding new one • Consistency ie. track changes and revisions consistently for code, config files and binary dependencies
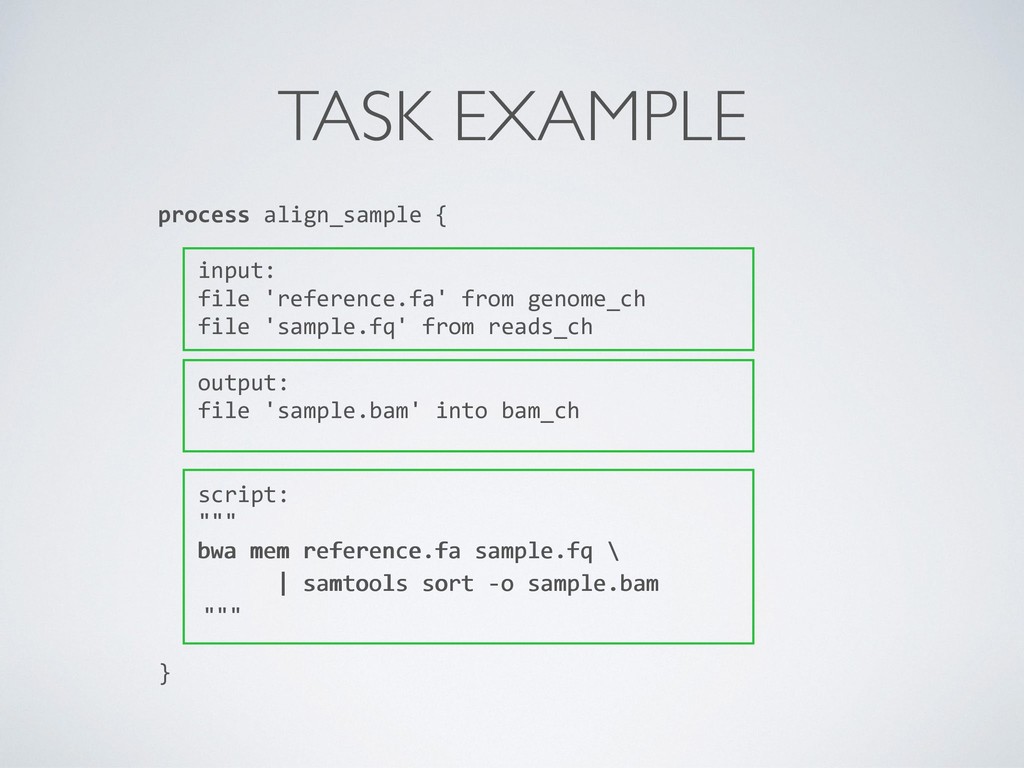
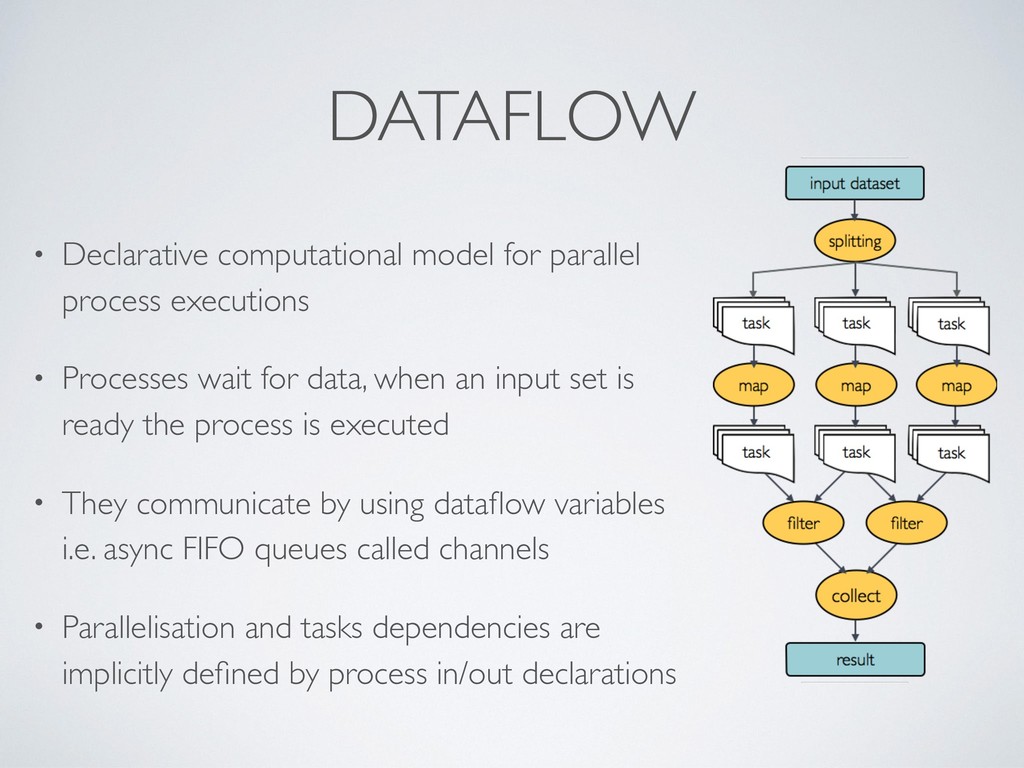
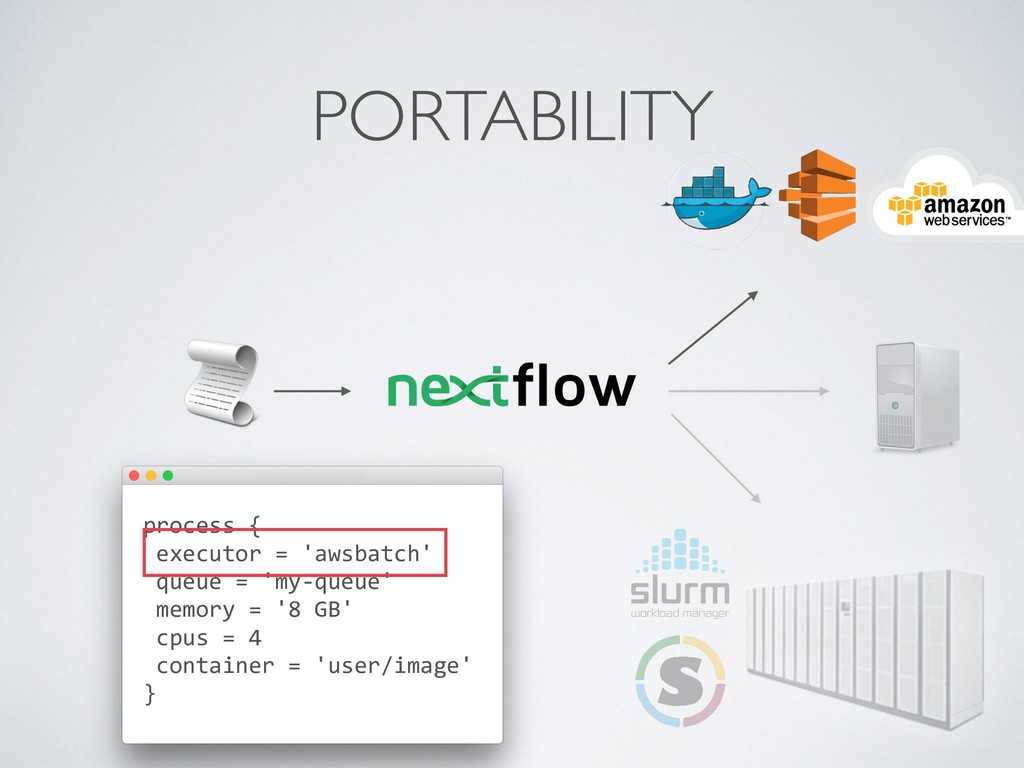
composition, simplifies most use cases + general purpose programming lang. for corner cases • Easy parallelisation 㱺 declarative reactive programming model based on dataflow paradigm, implicit portable parallelism • Self-contained 㱺 functional approach, a task execution is idempotent ie. cannot modify the state of other tasks + isolate dependencies with containers • Portable deployments 㱺 executor abstraction layer + deployment configuration from implementation logic
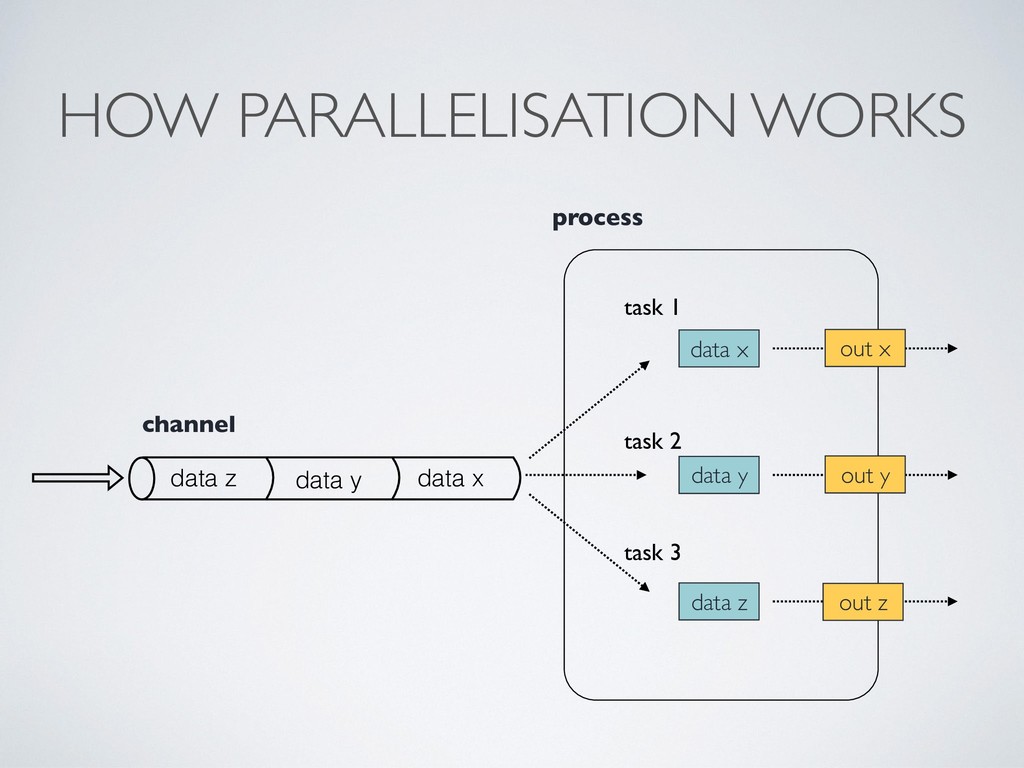
Processes wait for data, when an input set is ready the process is executed • They communicate by using dataflow variables i.e. async FIFO queues called channels • Parallelisation and tasks dependencies are implicitly defined by process in/out declarations
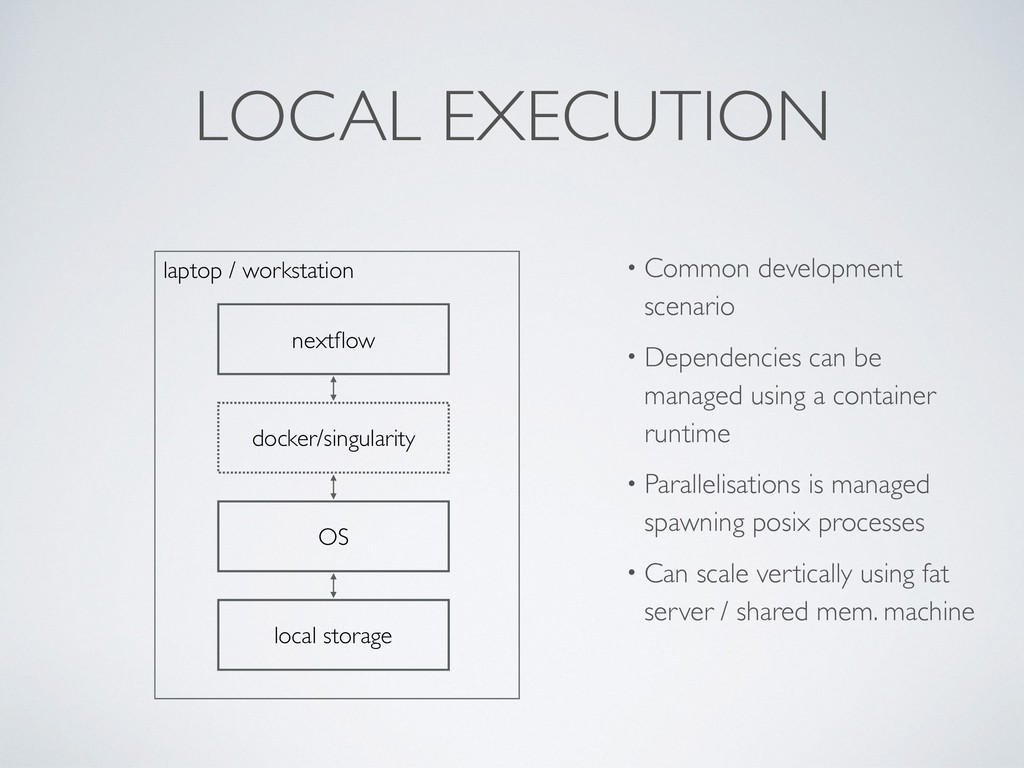
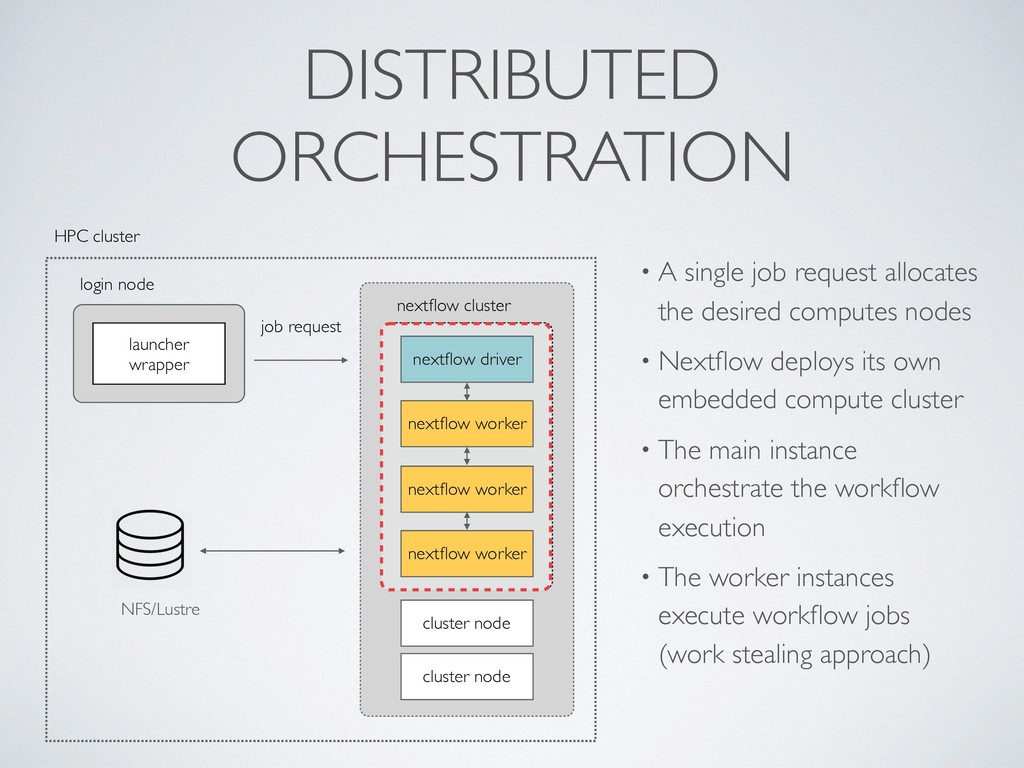
managed using a container runtime • Parallelisations is managed spawning posix processes • Can scale vertically using fat server / shared mem. machine nextflow OS local storage docker/singularity laptop / workstation
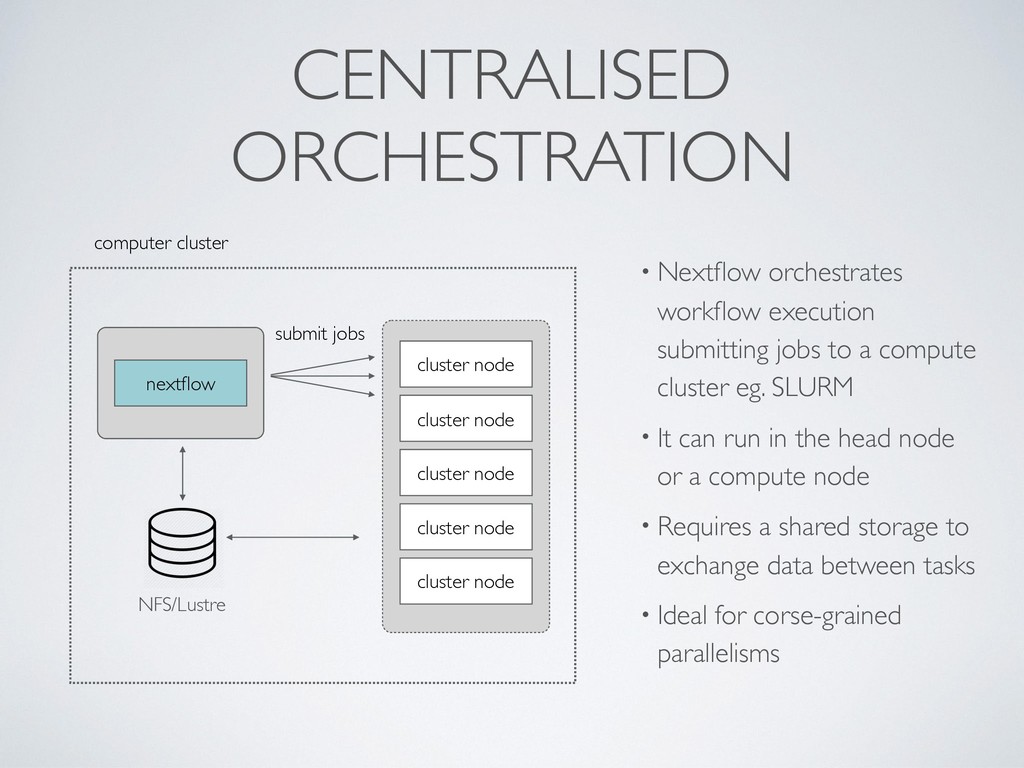
jobs to a compute cluster eg. SLURM • It can run in the head node or a compute node • Requires a shared storage to exchange data between tasks • Ideal for corse-grained parallelisms NFS/Lustre cluster node cluster node cluster node cluster node submit jobs cluster node nextflow
incorporate any tool w/o any extra adapter • Fine control over tasks parallelisation • Scalability 100㱺1M jobs • One liner installer • Suited for production workflows + bioinformaticians • Web based platform • Built-in integration with many tools and dataset • Little control over tasks parallelisation • Scalability 10㱺1K jobs • Complex installation and maintenance • Suited for training + less experienced bioinformaticians
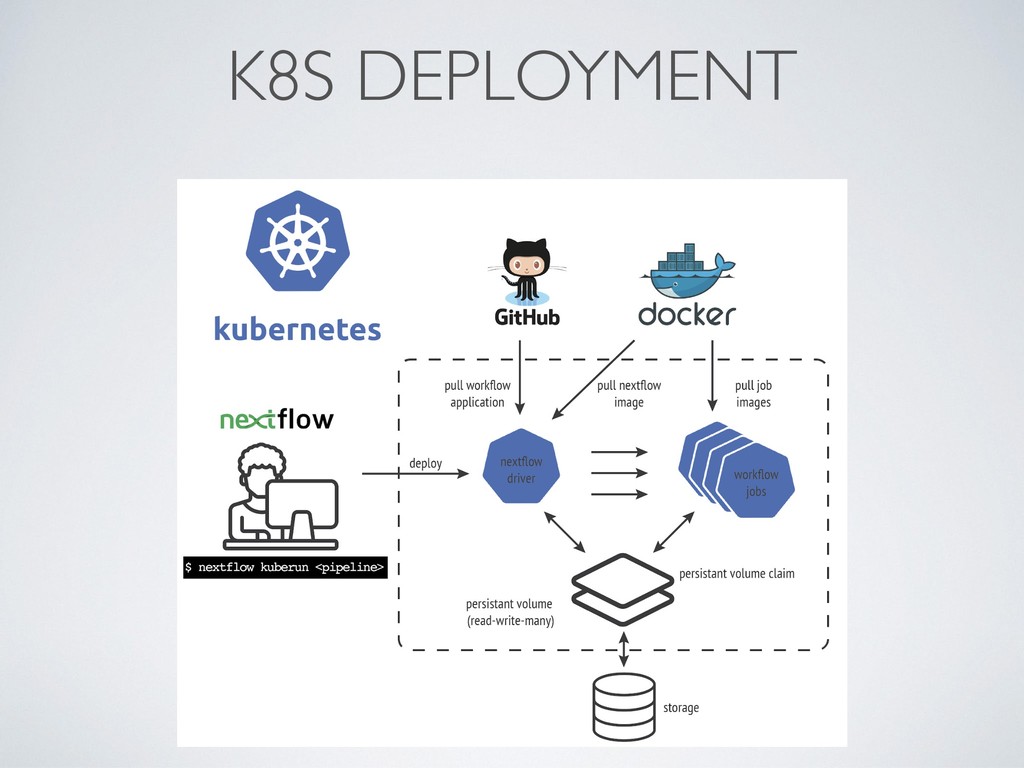
model • Can manage any data structure • Compute DAG at runtime • Built-in support for clusters and cloud • No (yet) support for sub-workflows • Built-in support for Git/GitHub, etc., manage pipeline revisions • Groovy/JVM based • Command line oriented tool • Pull model • Rules defined using file name patterns • Compute DAG ahead • Custom scripts for cluster deployments • Support for sub-workflows • No support for source code management system • Python based
on top of a general purpose programming lang. • Concise, fluent (at least try to be!) • Single implementation, quick iterations • Language specification • Declarative meta- language (YAML/JSON) • Verbose • Many vendors/ implementations (and specification version)
startup: ms/secs vs minutes • Virtualise a process/application instead of a OS/Hardware • Immutable: don't change over time, thus guarantee replicability over executions. • Composable: the output of one container is directly consumable as input by another container. • Transparent: they are created with a well defined automated procedure.
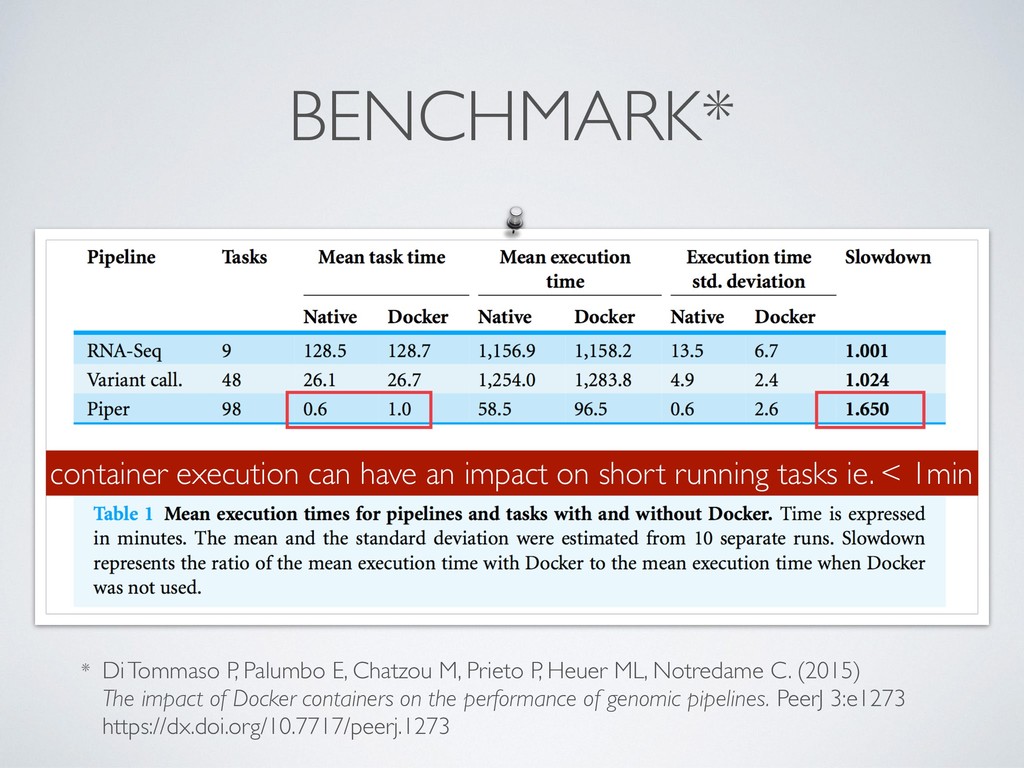
P, Heuer ML, Notredame C. (2015) The impact of Docker containers on the performance of genomic pipelines. PeerJ 3:e1273 https://dx.doi.org/10.7717/peerj.1273 container execution can have an impact on short running tasks ie. < 1min
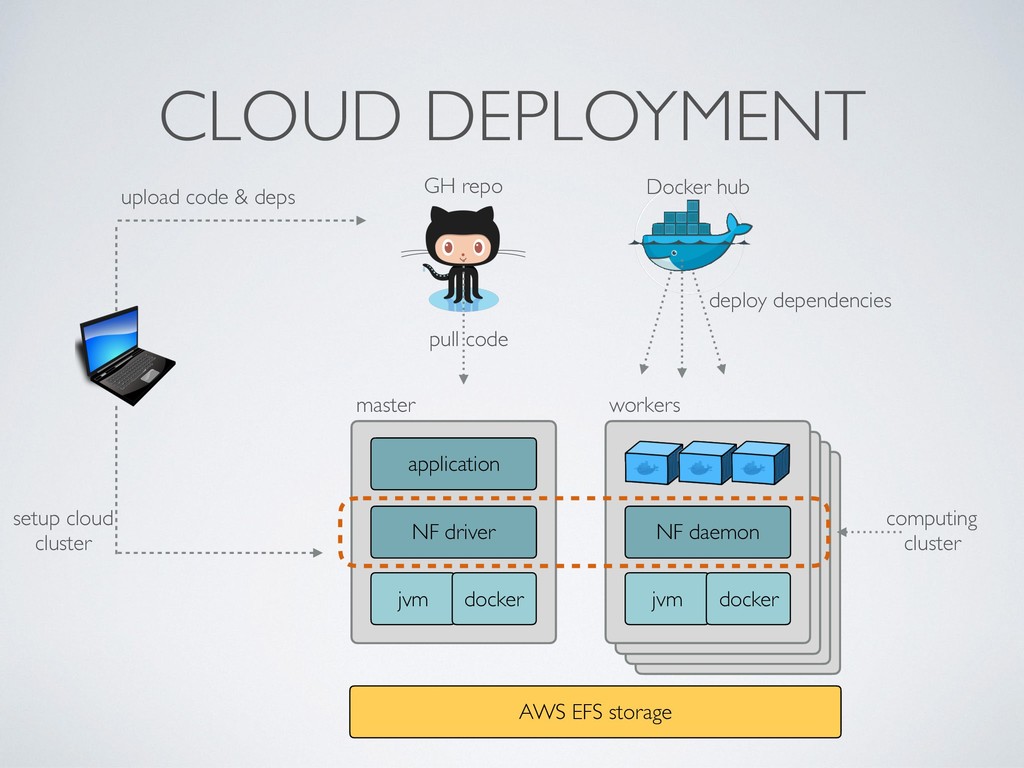
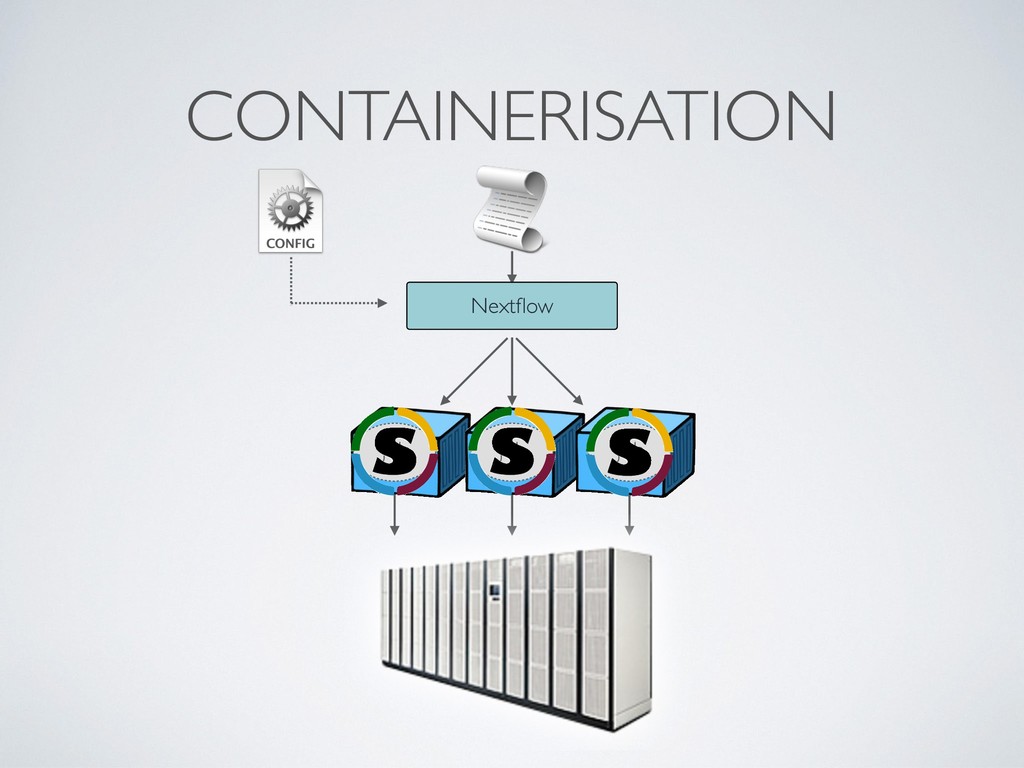
local deployment environment • Provides a reproducibles sandbox for third party users • Binary images preserve against software decay • Make it transparent ie. always include the Dockerfile • Docker image format is de-facto standard, it can be executed by different runtime eg. Singularity, Shifter, uDocker, etc.
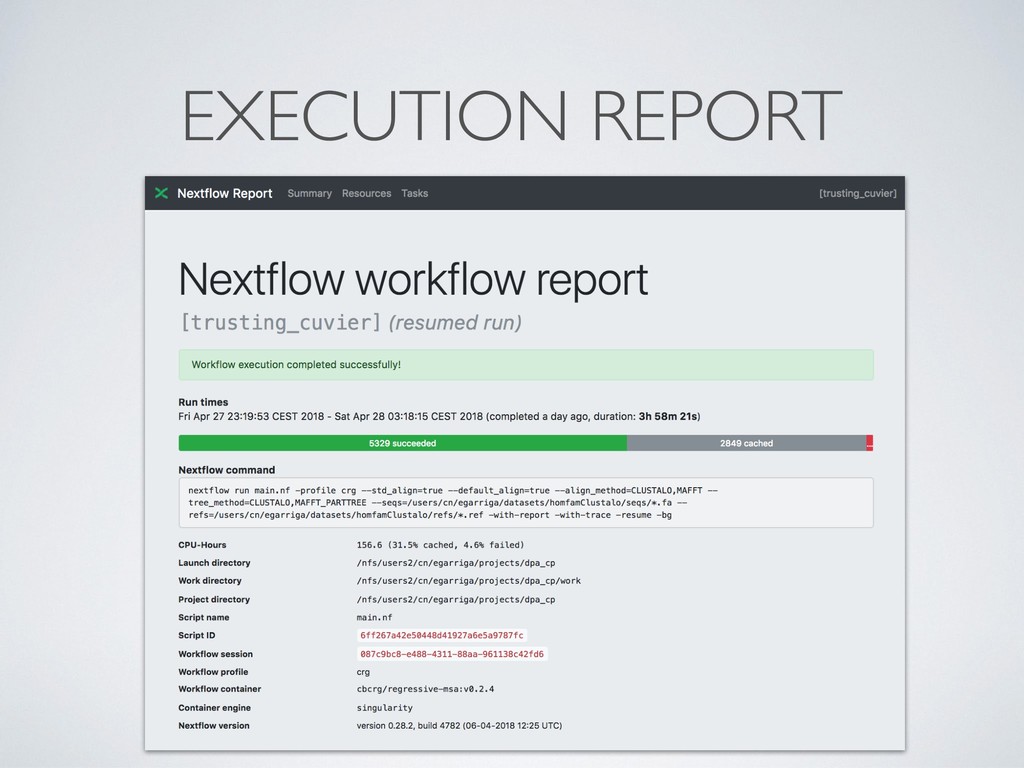
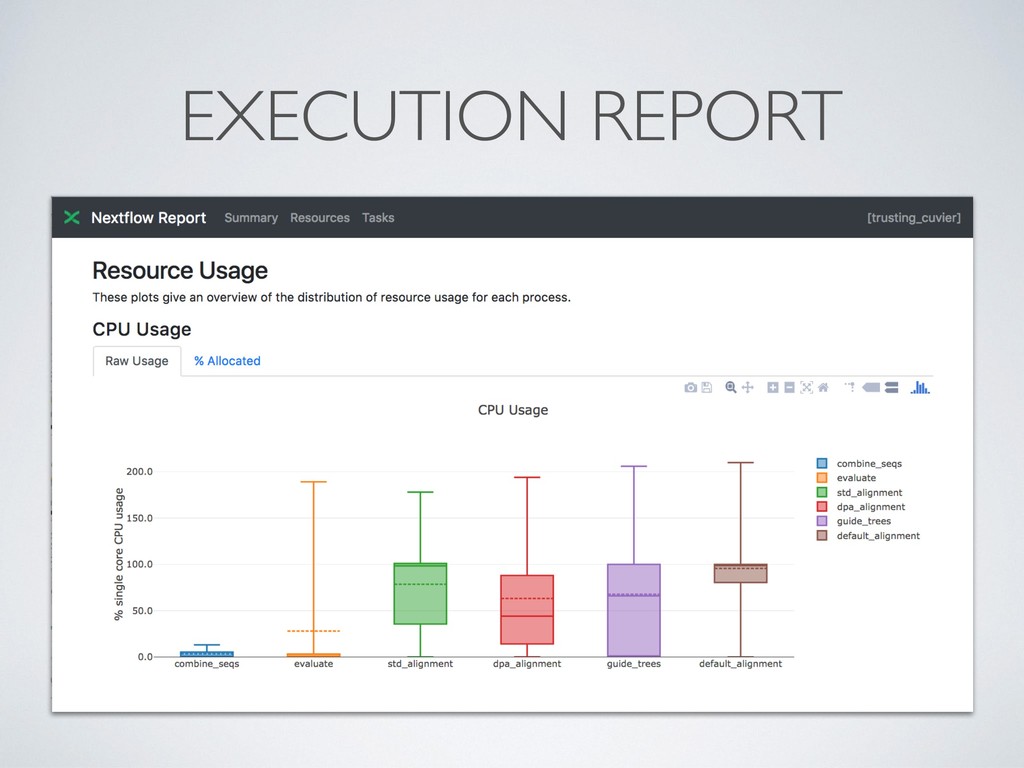
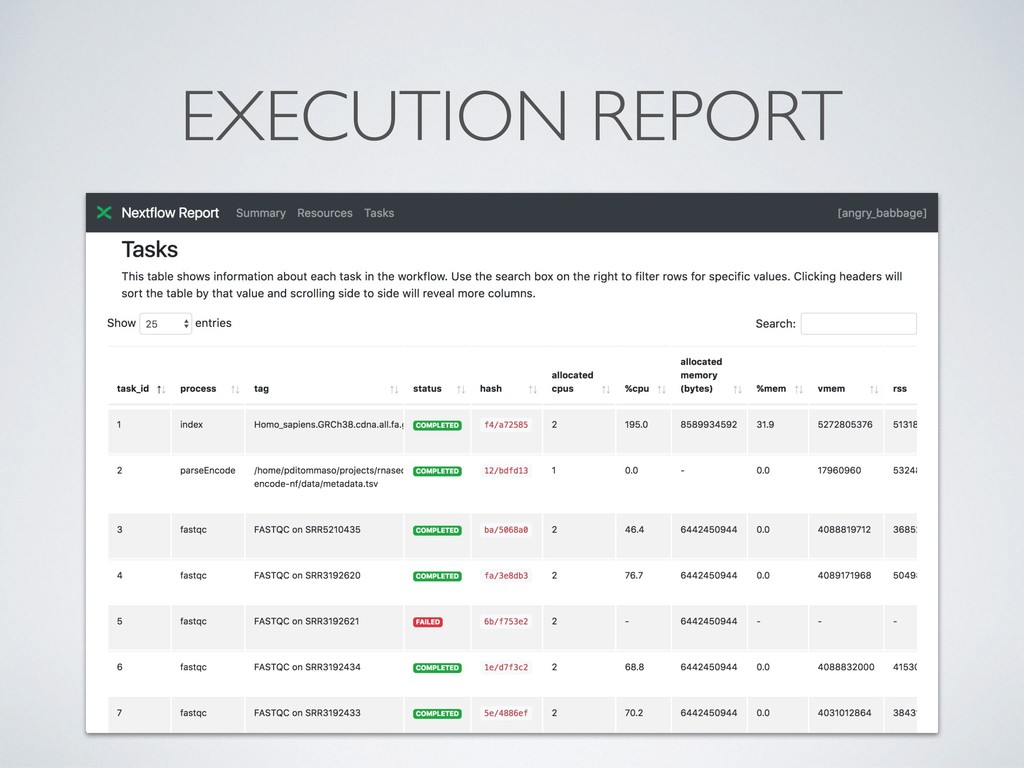
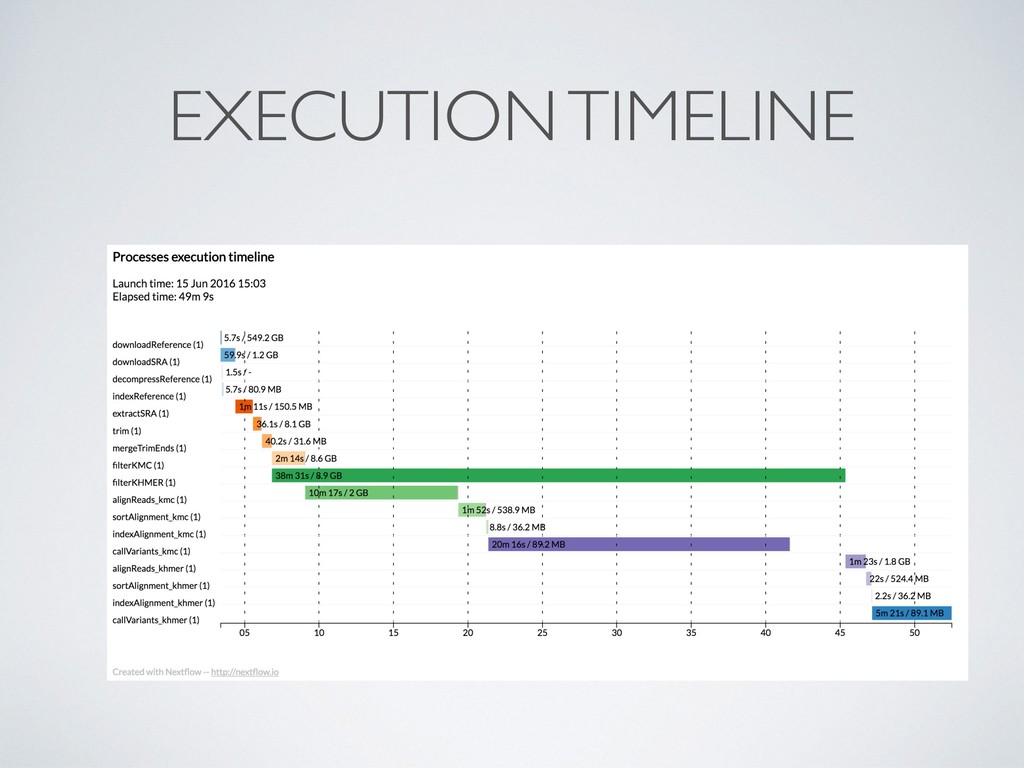
separate directory • This allows to safely record interrupted executions discarding • Dramatically simplify debugging ! • Computing resources can be defined in a *dynamic* manner, so that a failing task can be automatically re- execute with more memory, longer timeout, etc.
with Nextflow • Initially supported by SciLifeLab, QBiC and A*Star Genome Institute Singapore • https://nf-co.re Alexander Peltzer Phil Ewels Andreas Wilm Maxime Garcia + others
Uniform access to local and remote big-data repositories such as Athena, DynamoDB, BigQuery, etc • Query result is mapped to Nextflow channel structure triggering process executions
execution model • Allow hybrid Nextflow and Spark applications • Mix the best of the two worlds, Nextflow for legacy tools/corse grain parallelisation and Spark for fine grain/distributed execution eg. GATK4
Task Execution API (working prototype) • WES: Workflow Execution API • Enable interoperability with GA4GH complaint platforms eg. Cancer Genomics Cloud and Broad FireCloud
underestimated. • Nextflow does not provide a magic solution but enables best-practices and provide support for community and industry standards. • It strictly separates the application logic from the configuration and deployment logic, enabling self-contained workflows. • Applications can be easily deployed across different environment in a reproducible manner with a single command. • The functional/reactive model allows applications to scale to millions of jobs with ease.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![HANDLING FILE PAIRS Channel.fromFilePairs("*_{1,2}.fq") ( gut, [gut_1.fq, gut_2.fq] ) (](https://files.speakerdeck.com/presentations/92d5fe3825ae421ea4a74672008f4698/slide_22.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}