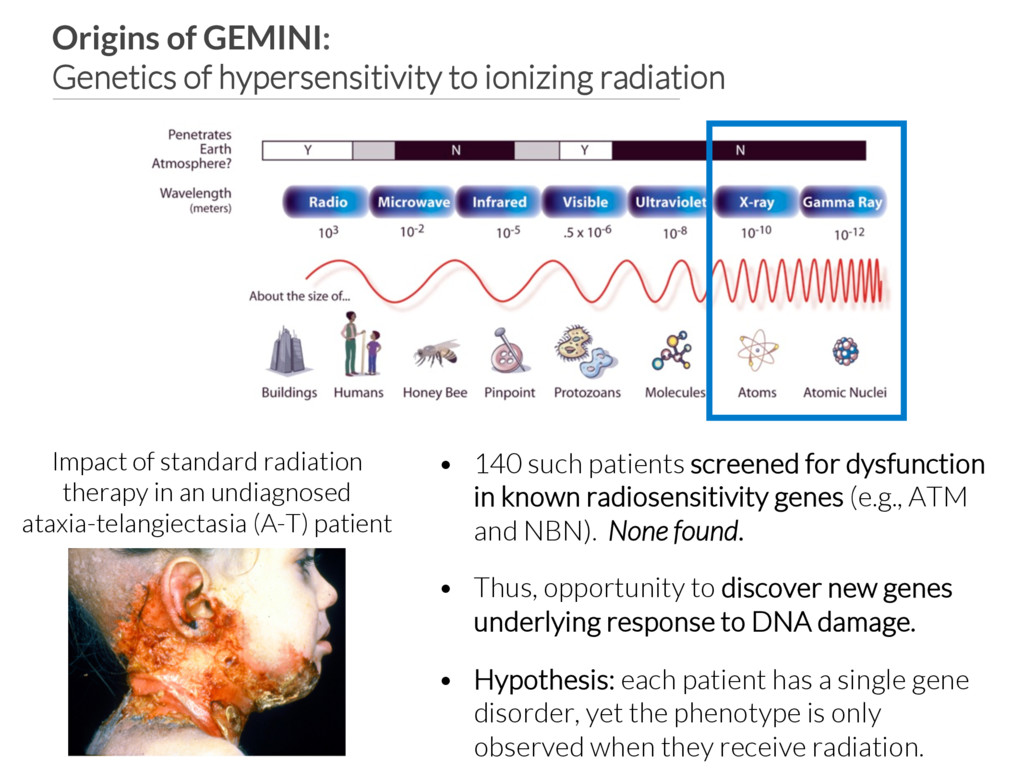
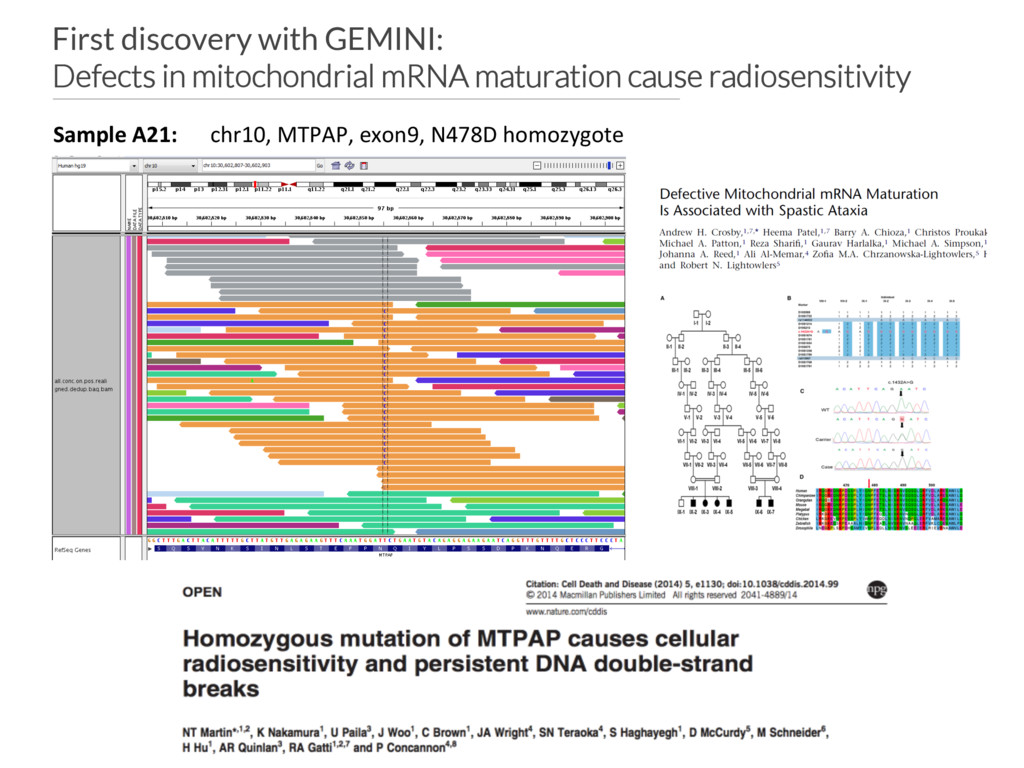
of standard radiation therapy in an undiagnosed ataxia-telangiectasia (A-T) patient • 140 such patients screened for dysfunction in known radiosensitivity genes (e.g., ATM and NBN). None found. • Thus, opportunity to discover new genes underlying response to DNA damage. • Hypothesis: each patient has a single gene disorder, yet the phenotype is only observed when they receive radiation.
FROM variants WHERE impact_severity == ‘HIGH’ AND max_aaf_all <= 0.001” --gt-filter “(gt_types).(LDL > 300).(!=HOM_REF).(count > 100) and (gt_types).(LDL < 100).(!=HOM_REF).(count < 10)" Which rare, deleterious variants are enriched in people with high LDL (>300 mg/dL) levels? gemini -q "SELECT * FROM variants WHERE impact_severity == ‘HIGH’ AND max_aaf_all <= 0.001” --gt-filter “(gt_types).(breed=“angus”).(!=HOM_REF).(count > 100) and (gt_types).(breed=“belgian”).(!=HOM_REF).(count < 10)" Which rare, deleterious variants are enriched in Angus cattle but not Belgian Blue? (theoretical at the moment)
ALT QUAL FILTER 2 41647 . A G 4495.41 PASS 2 45895 . A G 463.75 PASS 2 224970 . C T 4241.64 PASS 2 229934 . A G 5037.95 PASS 2 234130 . T G 3958 PASS 2 242732 . T TAAC 3193.19 PASS 2 242800 . T C 3929.77 PASS 2 243504 . C T 6628.06 PASS 2 243567 . T TA 3398.03 HRunFilter 2 262553 . T C 3503.49 PASS 2 264895 . G C 3774.13 PASS 2 269352 . G A 9802.28 PASS 2 276942 . A G 5878.58 PASS 2 277250 . G A 7051.35 PASS 2 279705 . C T 7139.54 PASS 2 283231 . A AT 6976.81 HRunFilter 2 675831 . G T 865.05 PASS 2 676177 . C G 4961.19 PASS 2 905368 . C G 101.98 ABFilter; 2 905369 . C G 28.97 ABFilter 2 905393 . C G 930.81 QDFilter 2 905427 . C G 140.17 QDFilter 2 905442 . A T 131.51 ABFilter 2 905492 . T G 550.3 QDFilter 2 905494 . C G 48.5 ABFilter 2 905533 . C T 320.33 ABFilter 2 905576 . T G 72.09 QDFilter 2 905581 . C T 1276.63 QDFilter 2 905595 . G C 390.15 ABFilter 2 905634 . A C 393.91 QDFilter 2 905687 . C G 3233.06 ABFilter 2 905736 . A T 1324.63 QDFilter 2 905763 . G C 15.12 ABFilter Tabix’ed . . .
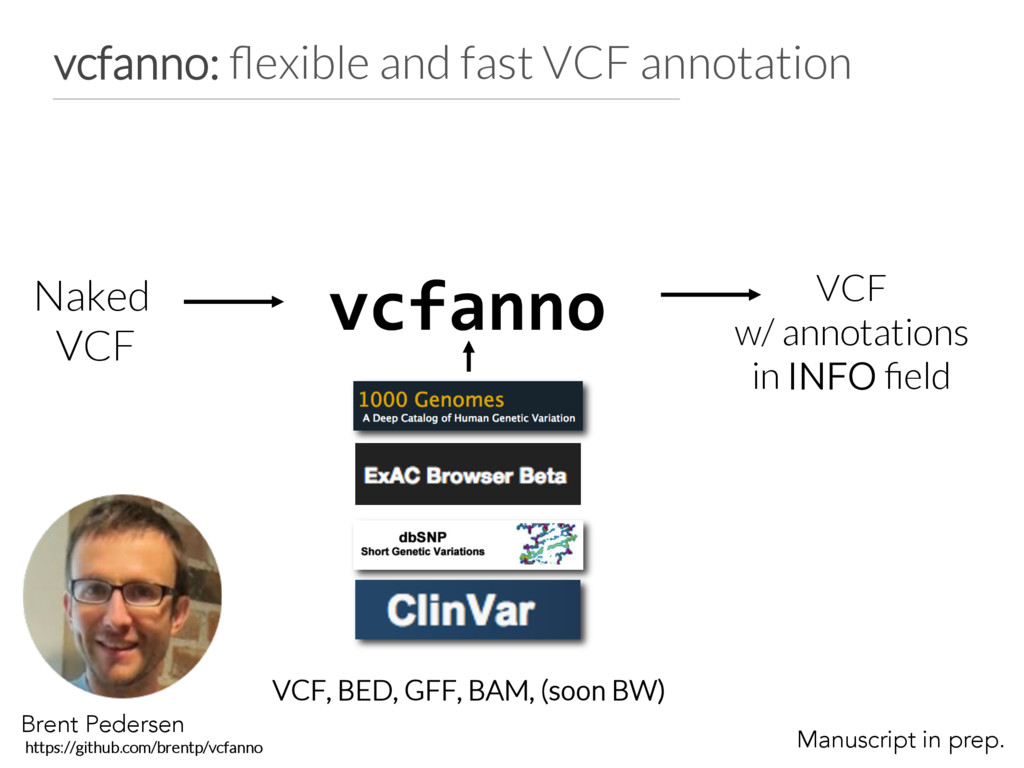
fields=["ID"] names=["rs_ids"] ops=[“concat"] [[annotation]] file="gerp.elements.bed.gz" columns=[4,4] names=[“gerp_mean”,”gerp_var”] ops=[“mean”, "lua:variance(vals)"] vcfanno configuration file. Allows multiple annotations from each file Can rename the annotations in the resulting VCF Multiple operations to summarize the results of multiple hits in annot. file: mean, max, min concat, count, uniq first, flag Match on POS+REF+ALT for VCF annotations. Lua for custom computations. variance() defined in custom.js
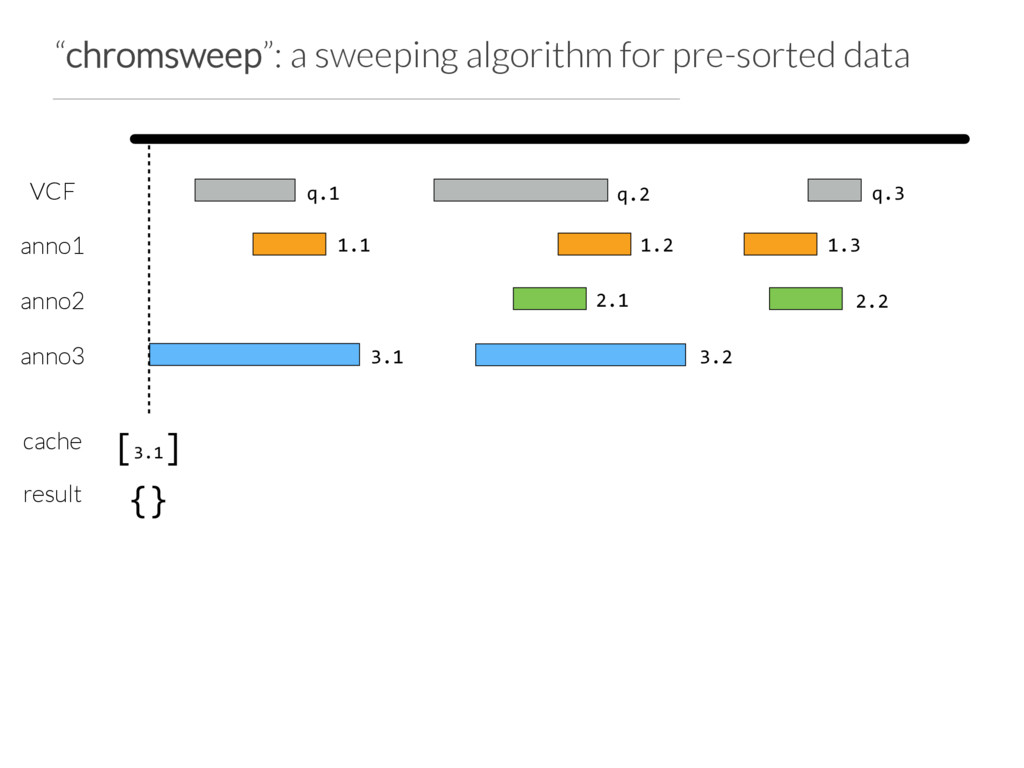
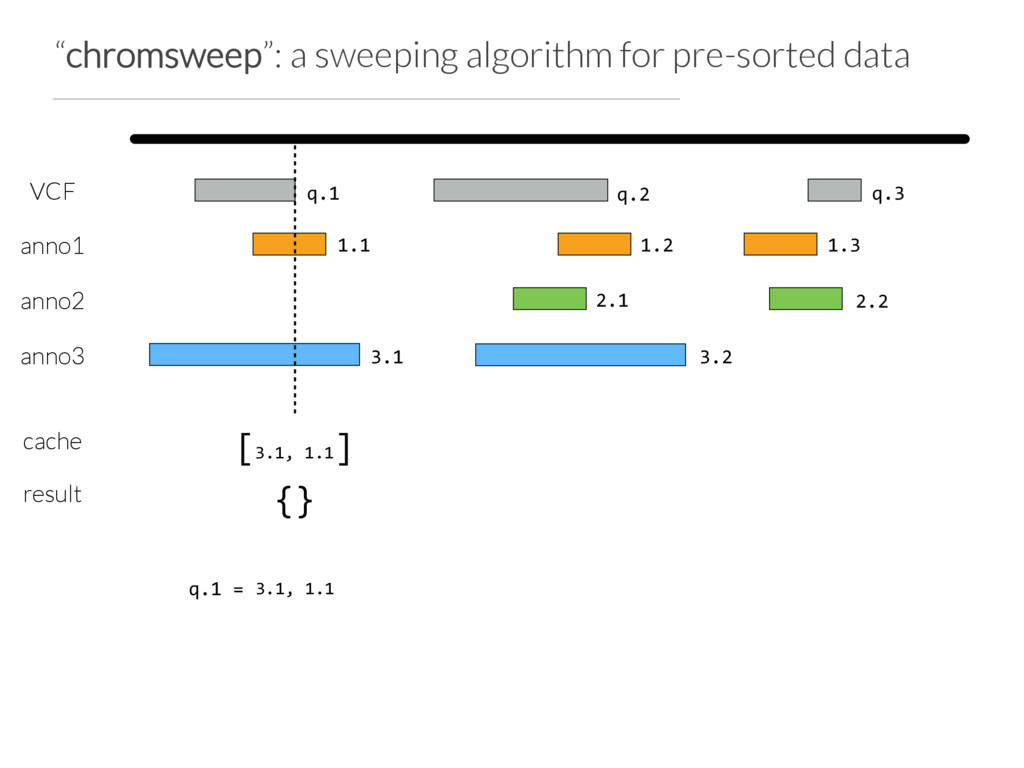
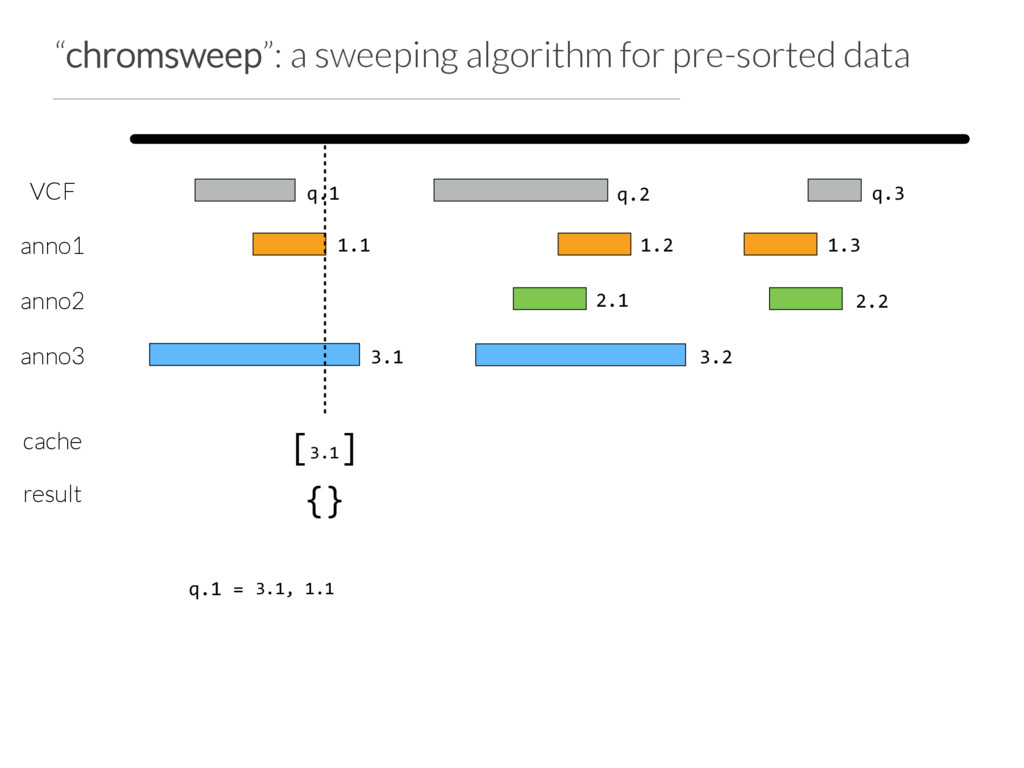
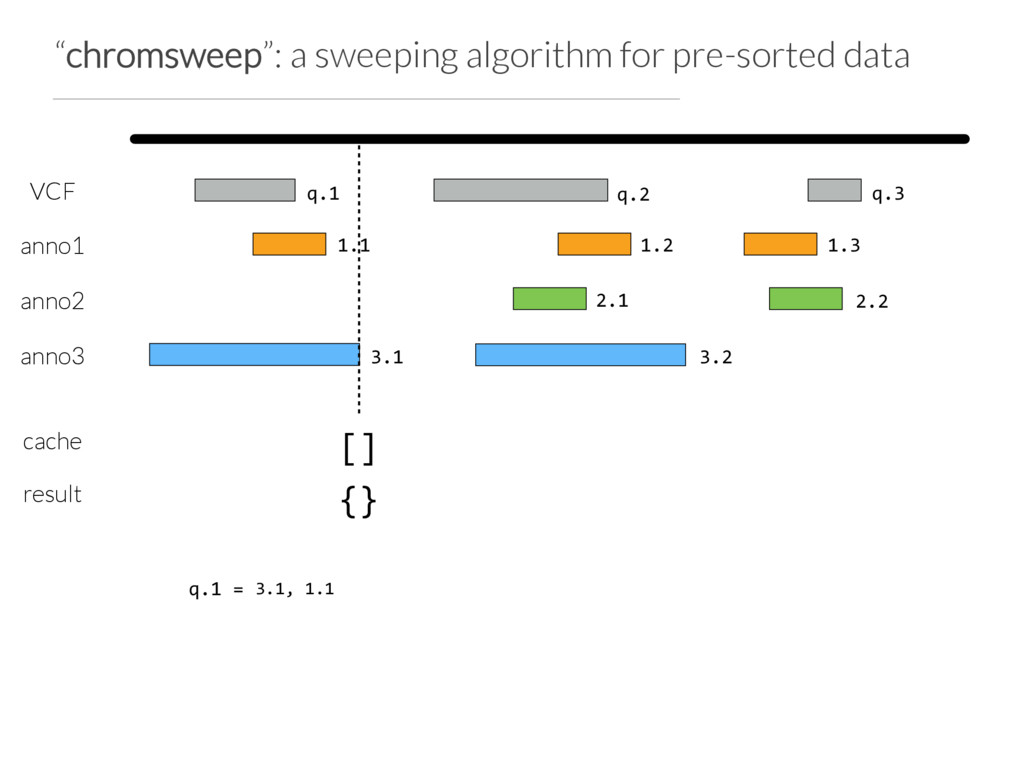
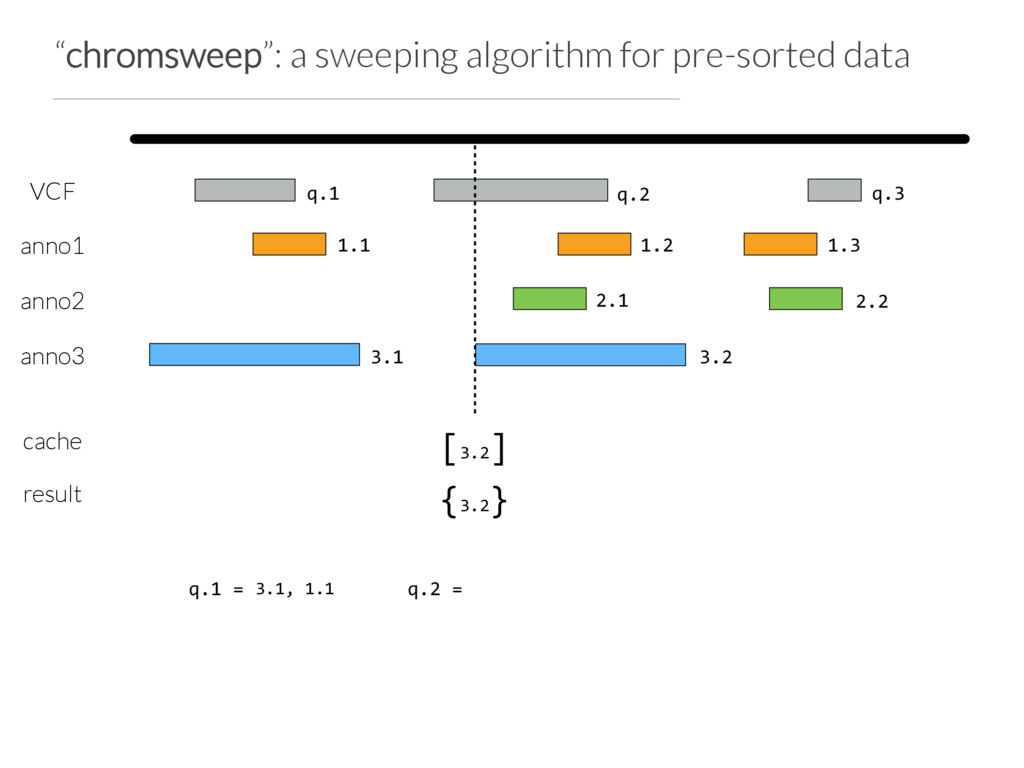
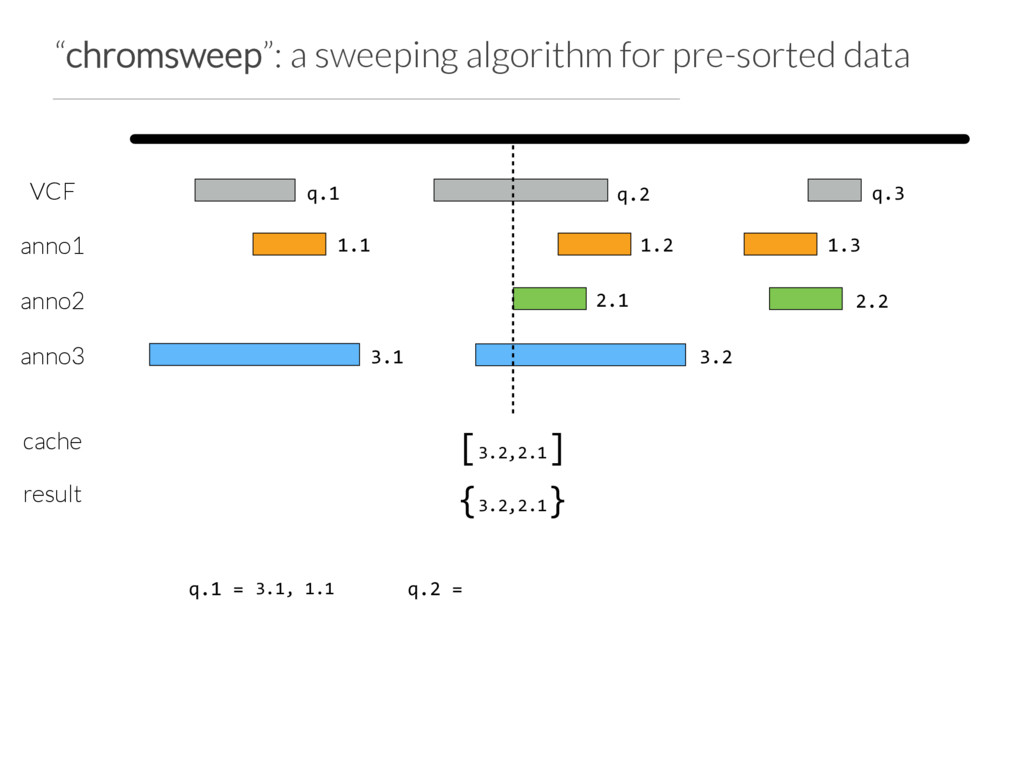
Step 1: partition the query set at “breaks” in the data or when N (e.g. 10) intervals are found Step 2: Use Tabix to extract the records germane to a chunk from each annotation file Step 3: Chromsweep each chunk independently.
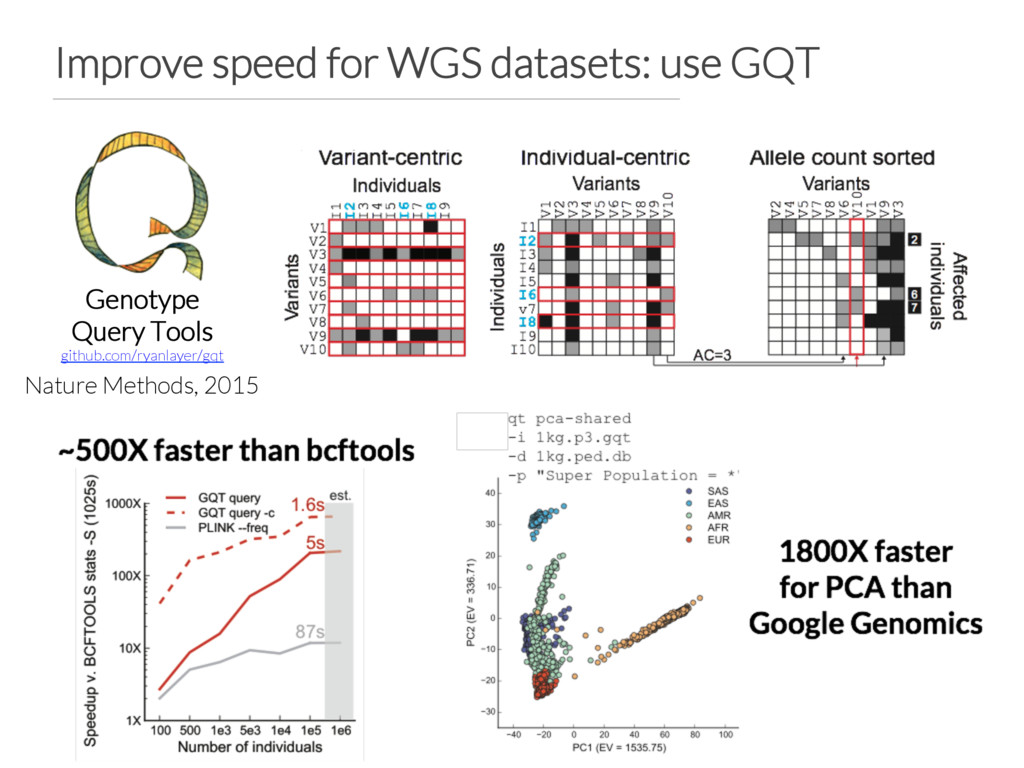
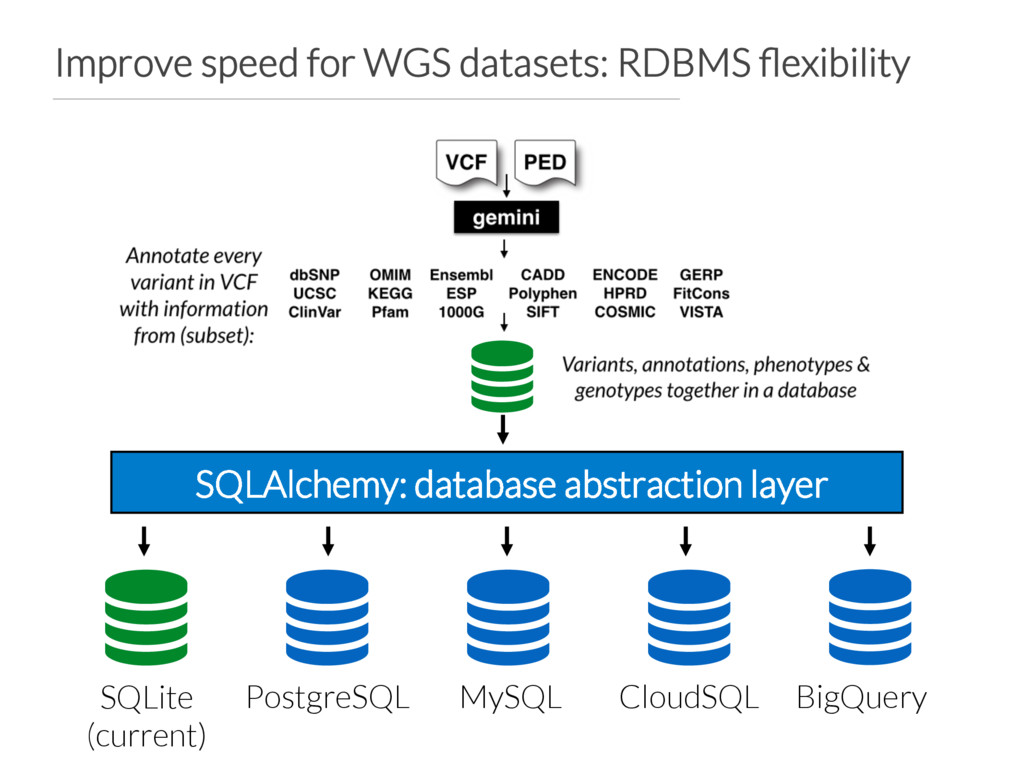
build Vcfanno configuration file points to appropriate annotations GEMINI database is created based on vcfanno configuration file GEMINI database creation should be ~60X faster How do we support other species?
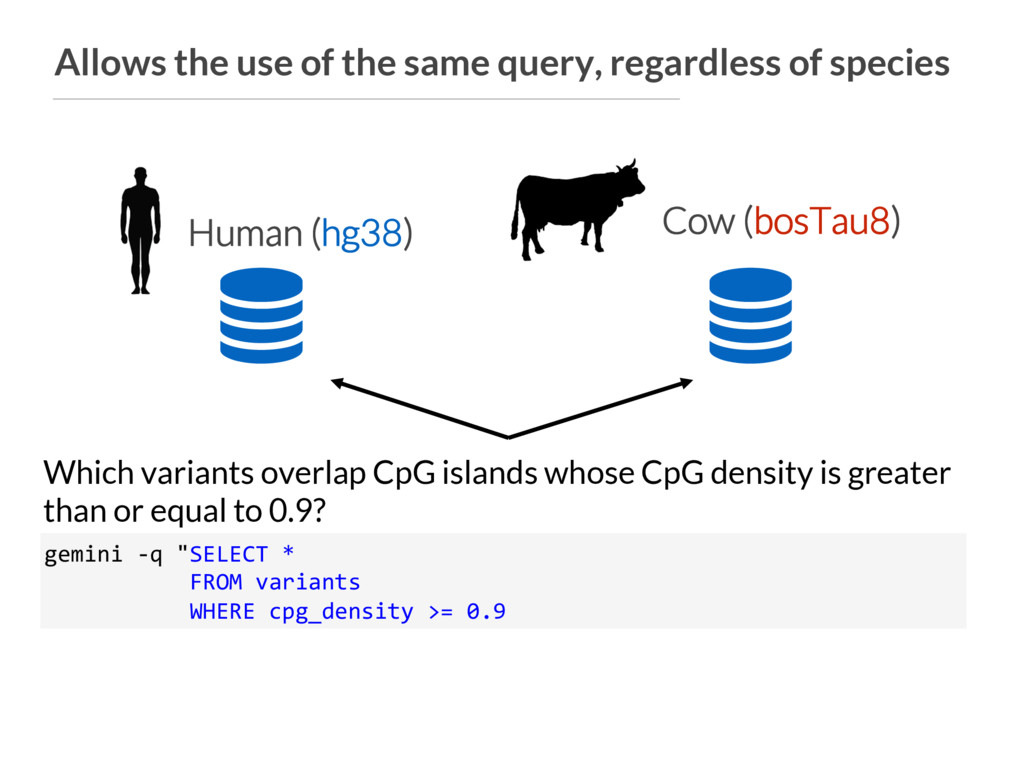
gemini -q "SELECT * FROM variants WHERE cpg_density >= 0.9 Which variants overlap CpG islands whose CpG density is greater than or equal to 0.9? Human (hg38) Cow (bosTau8)
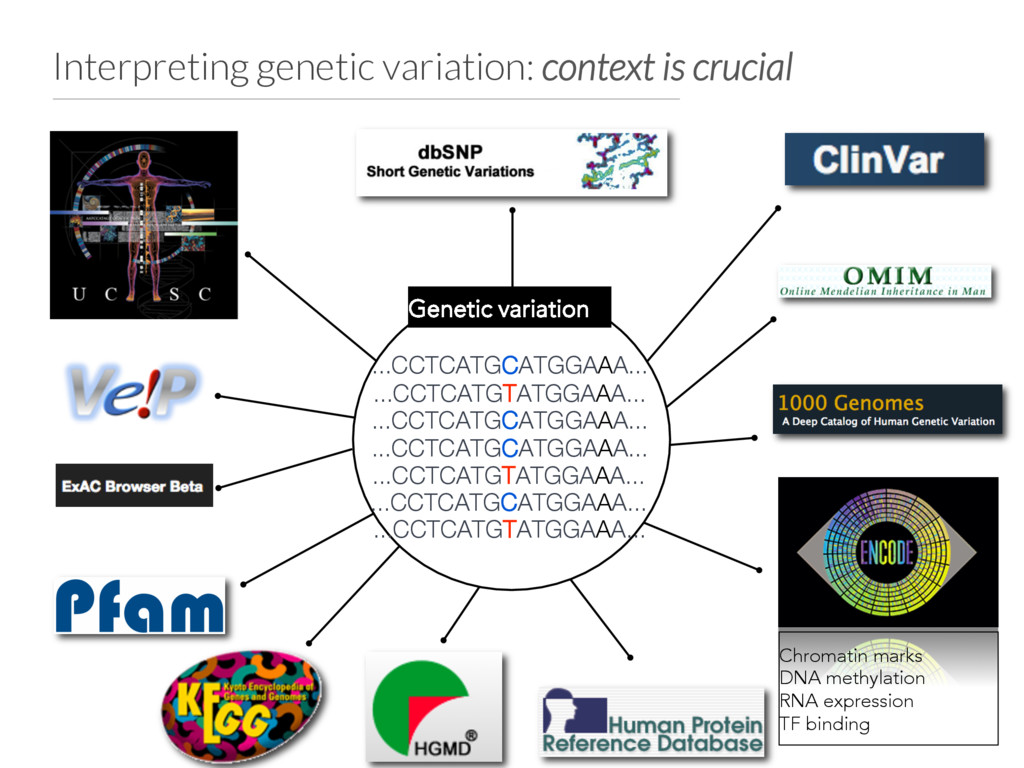
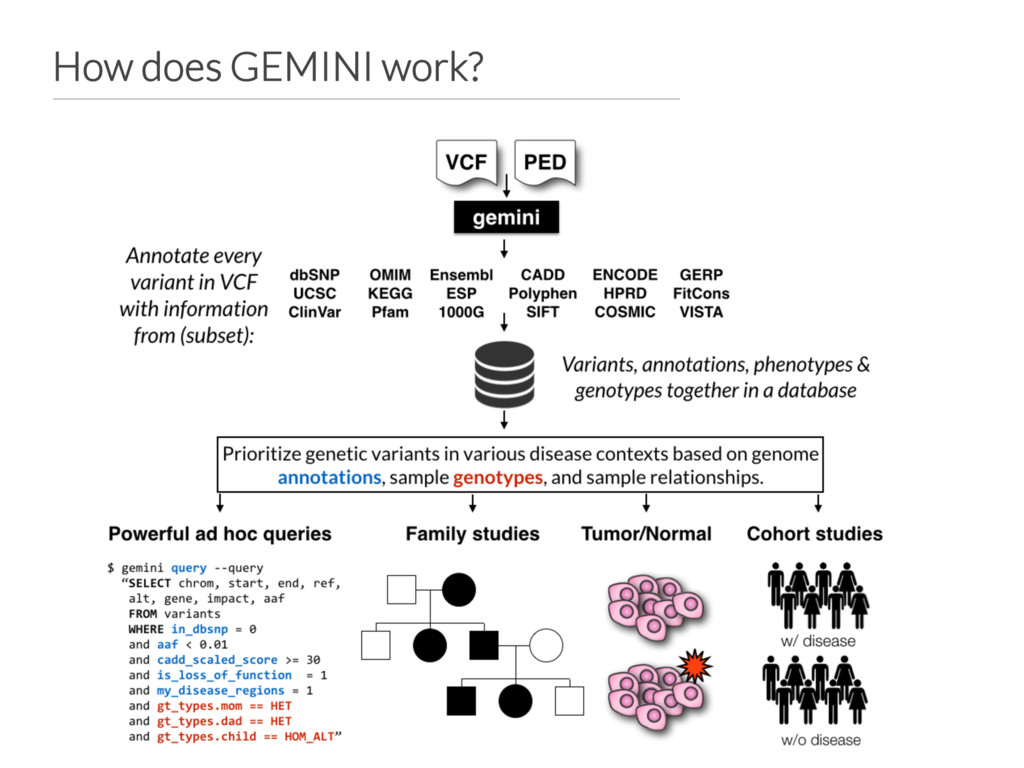
variation from WES and WGS studies. • Integrates variants, genotypes, phenotypes and annotations into a simple database. • Current focus: • Improving scalability for WGS • Support for any (diploid) species • Expected release: April 2016 github.com/arq5x/gemini gemini.rtfd.org
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[[annotation]] file=“ExAC.v3.vcf” fields=[“AF”, “AC_Het”] names=[“exac_aaf”, “exac_num_het”] ops=[“first”, “first”] [[annotation]] file="dbsnp.b141.vcf.gz"](https://files.speakerdeck.com/presentations/4ce5c16bbf394a8bb233b705d86d415b/slide_14.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[[annotation]] file=“cpg.hg38.bed.gz" fields=[4] names=[“cpg_density"] ops=[“mean"] [[annotation]] file=“rmsk.hg38.bed.gz" fields=[4] names=[“rmsk"] ops=[“concat”]](https://files.speakerdeck.com/presentations/4ce5c16bbf394a8bb233b705d86d415b/slide_35.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}