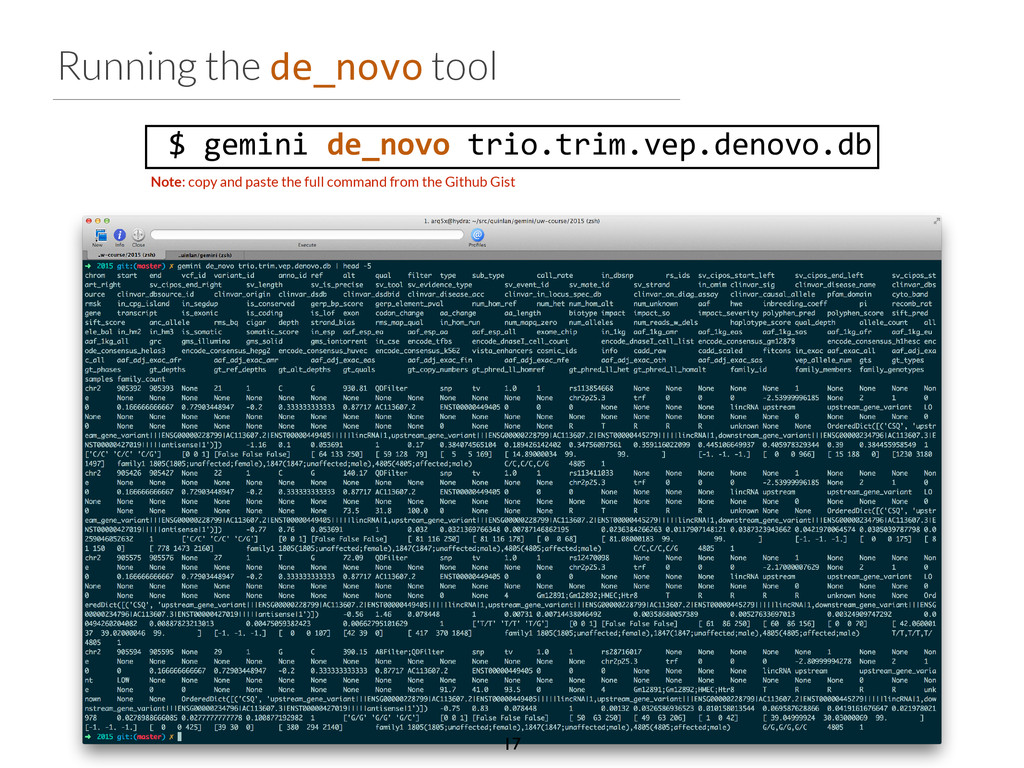
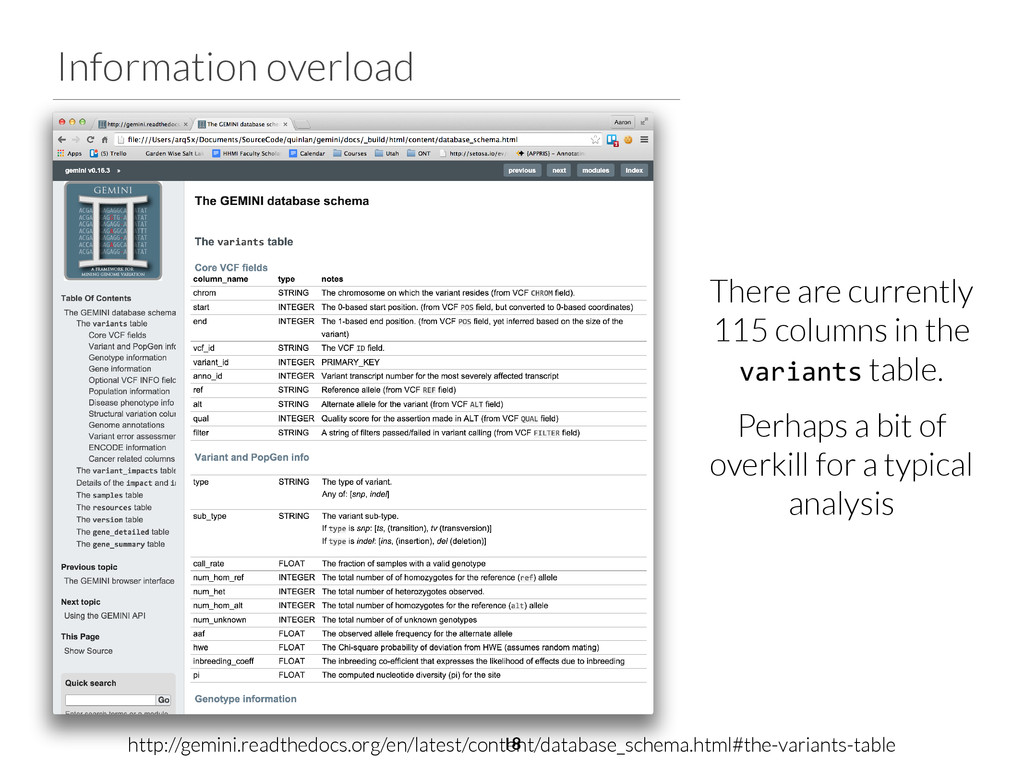
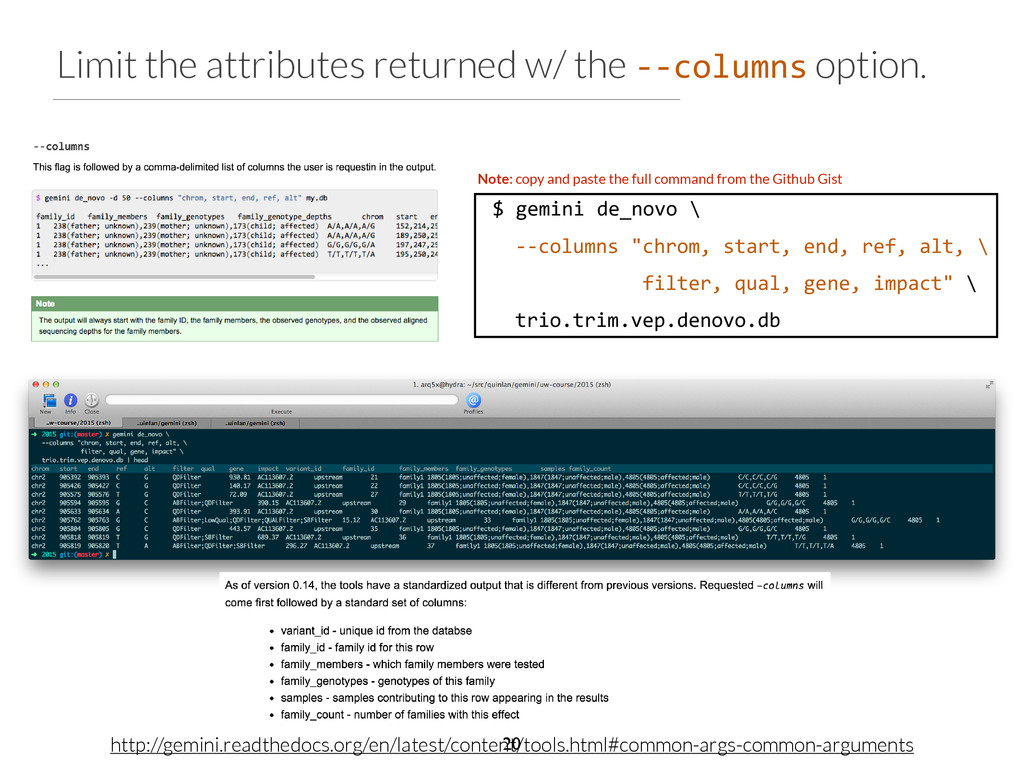
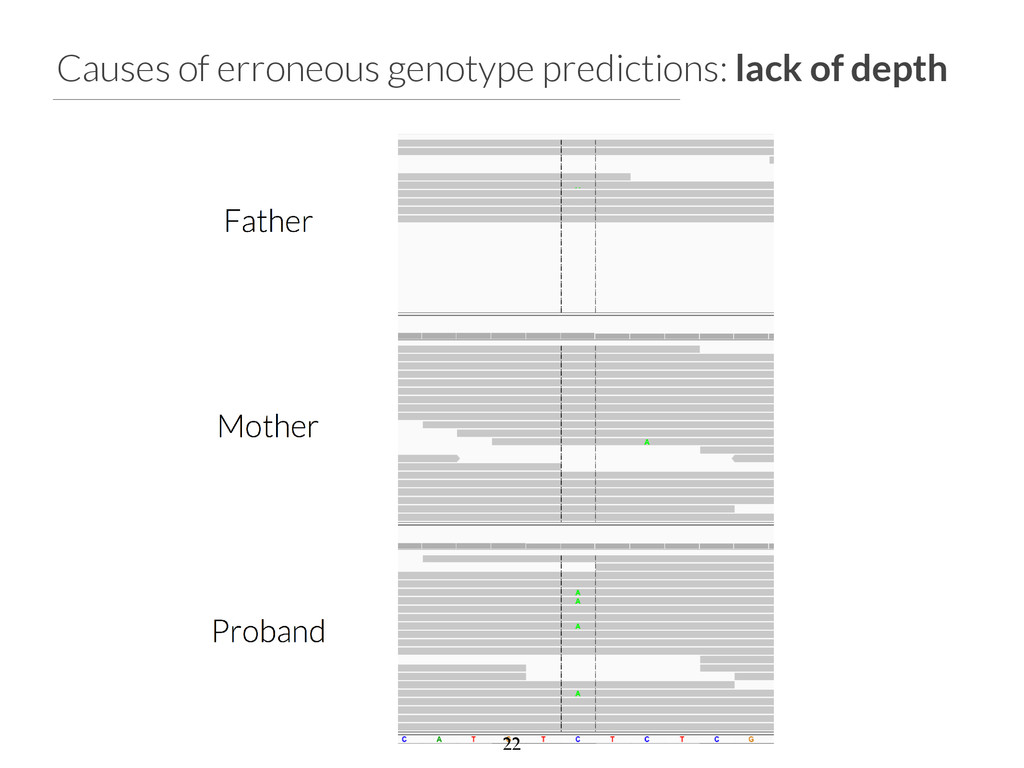
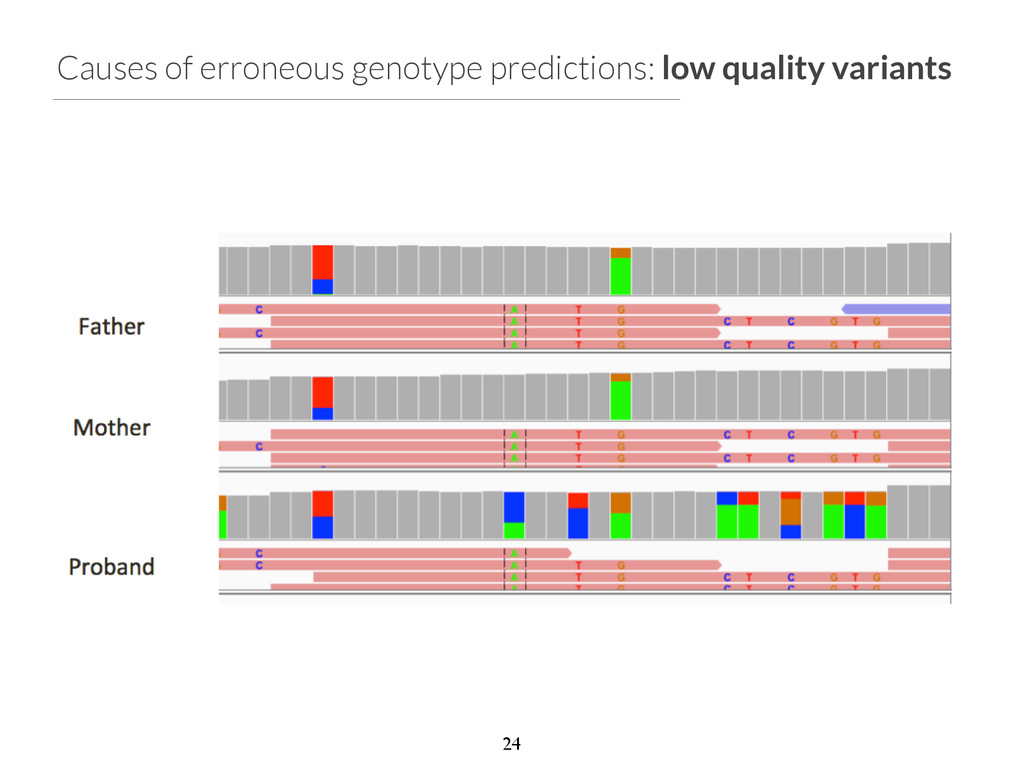
start end ref alt filter qual gene impact variant_id family_id family_members family_genotypes samples family_count chr2 96525735 96525736 T C None 1929.31 ANKRD36C non_syn_coding 2537 family1 1805,1847,4805 T/T,T/T,T/C 4805 1 chr2 96525749 96525750 T A None 1513.36 ANKRD36C non_syn_coding 2538 family1 1805,1847,4805 T/T,T/T,T/A 4805 1 chr2 96525754 96525755 A T None 1699.28 ANKRD36C non_syn_coding 2539 family1 1805,1847,4805 A/A,A/A,A/T 4805 1 chr15 41229630 41229631 T G None 2116.49 DLL4 non_syn_coding 7892 family1 1805,1847,4805 T/T,T/T,T/G 4805 1 chr17 55183812 55183813 A G None 2155.84 AKAP1 non_syn_coding 13311 family1 1805,1847,4805 A/A,A/A,A/G 4805 1 chr22 43027436 43027437 C T None 1320.03 CYB5R3 non_syn_coding 16718 family1 1805,1847,4805 C/C,C/C,C/T 4805 1 Phenotype: blue skin disease $ gemini de_novo \ -‐-‐columns "chrom, start, end, ref, alt, \ filter, qual, gene, impact" \ -‐d 15 \ -‐-‐filter "filter is NULL \ and is_coding = 1 and impact_severity != ‘LOW’ \ and (aaf_1kg_eur <= 0.005 or aaf_1kg_eur is NULL) \ and (aaf_esp_ea <= 0.005 or aaf_esp_ea is NULL)" \ trio.trim.vep.denovo.db Which gene can we rule out at a glance? 30
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}