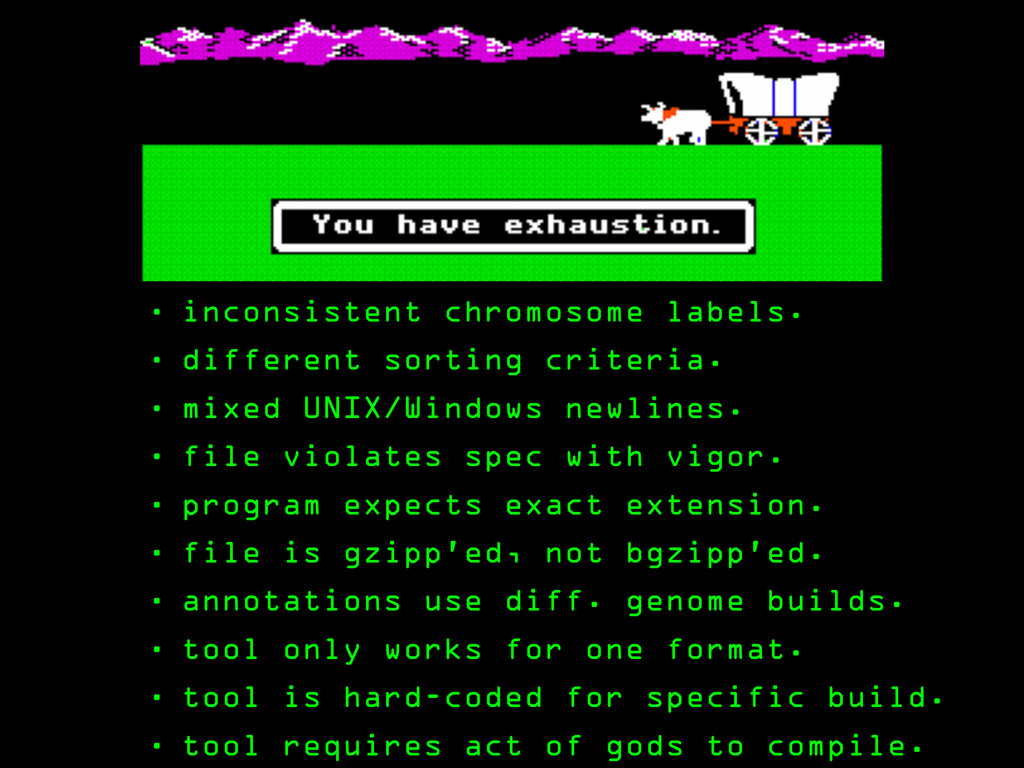
UNIX/Windows newlines. • file violates spec with vigor. • program expects exact extension. • file is gzipp’ed, not bgzipp’ed. • annotations use diff. genome builds. • tool only works for one format. • tool is hard-coded for specific build. • tool requires act of gods to compile.
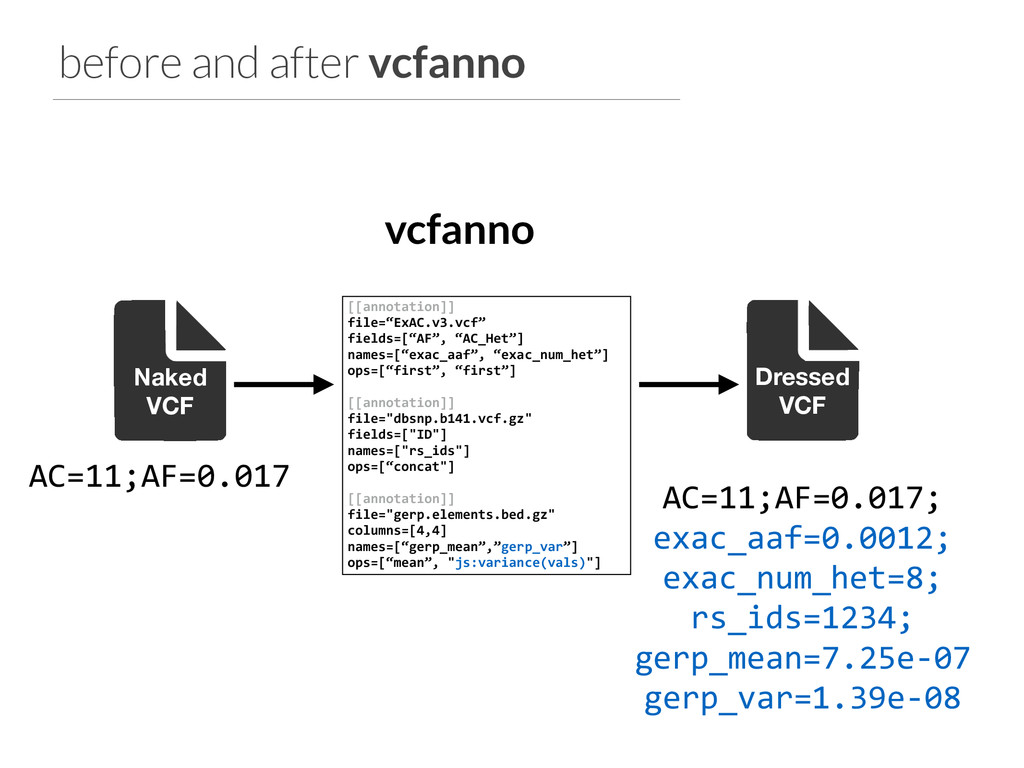
ops=[“first”, “first”] ! [[annotation]] file="dbsnp.b141.vcf.gz" fields=["ID"] names=["rs_ids"] ops=[“concat"] ! [[annotation]] file="gerp.elements.bed.gz" columns=[4,4] names=[“gerp_mean”,”gerp_var”] ops=[“mean”, "js:variance(vals)"] vcfanno configuration file. Allows multiple annotations from each file Can rename the annotations in the resulting VCF Match on POS+REF+ALT for VCF annotations.
ops=[“first”, “first”] ! [[annotation]] file="dbsnp.b141.vcf.gz" fields=["ID"] names=["rs_ids"] ops=[“concat"] ! [[annotation]] file="gerp.elements.bed.gz" columns=[4,4] names=[“gerp_mean”,”gerp_var”] ops=[“mean”, "js:variance(vals)"] vcfanno configuration file. Allows multiple annotations from each file Can rename the annotations in the resulting VCF Multiple operations to summarize the results of multiple hits in annot. file: mean, max, min concat, count, uniq first, flag Match on POS+REF+ALT for VCF annotations.
ops=[“first”, “first”] ! [[annotation]] file="dbsnp.b141.vcf.gz" fields=["ID"] names=["rs_ids"] ops=[“concat"] ! [[annotation]] file="gerp.elements.bed.gz" columns=[4,4] names=[“gerp_mean”,”gerp_var”] ops=[“mean”, "js:variance(vals)"] vcfanno configuration file. Allows multiple annotations from each file Can rename the annotations in the resulting VCF Multiple operations to summarize the results of multiple hits in annot. file: mean, max, min concat, count, uniq first, flag Match on POS+REF+ALT for VCF annotations. Javascript for custom computations. variance() defined in custom.js
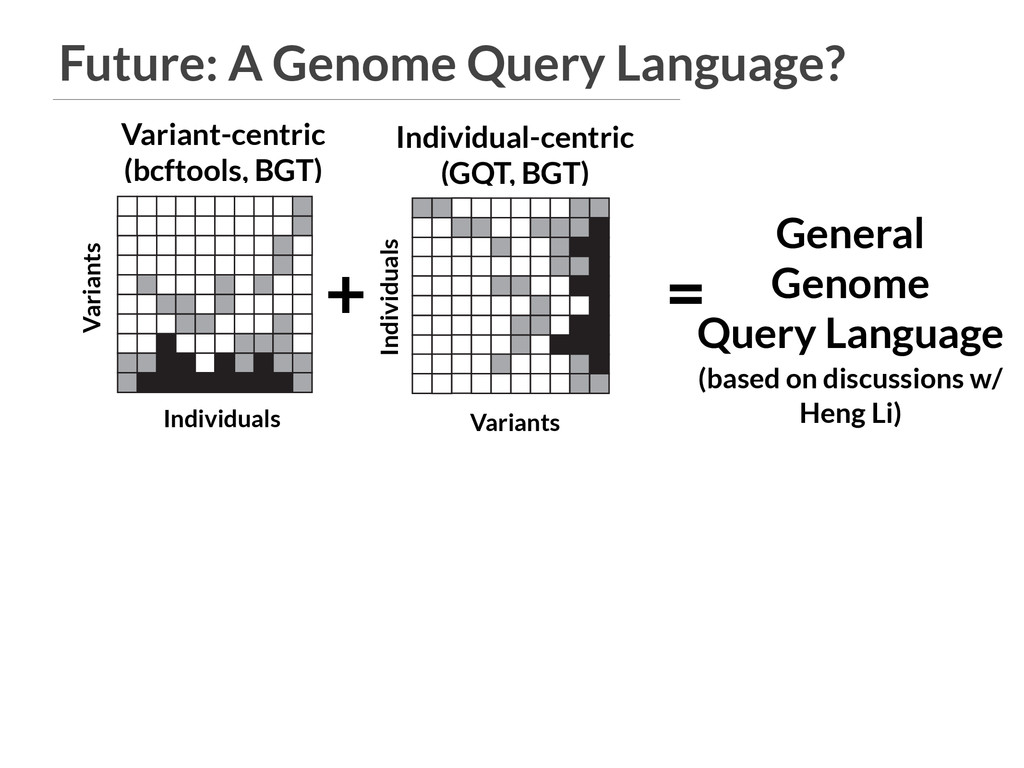
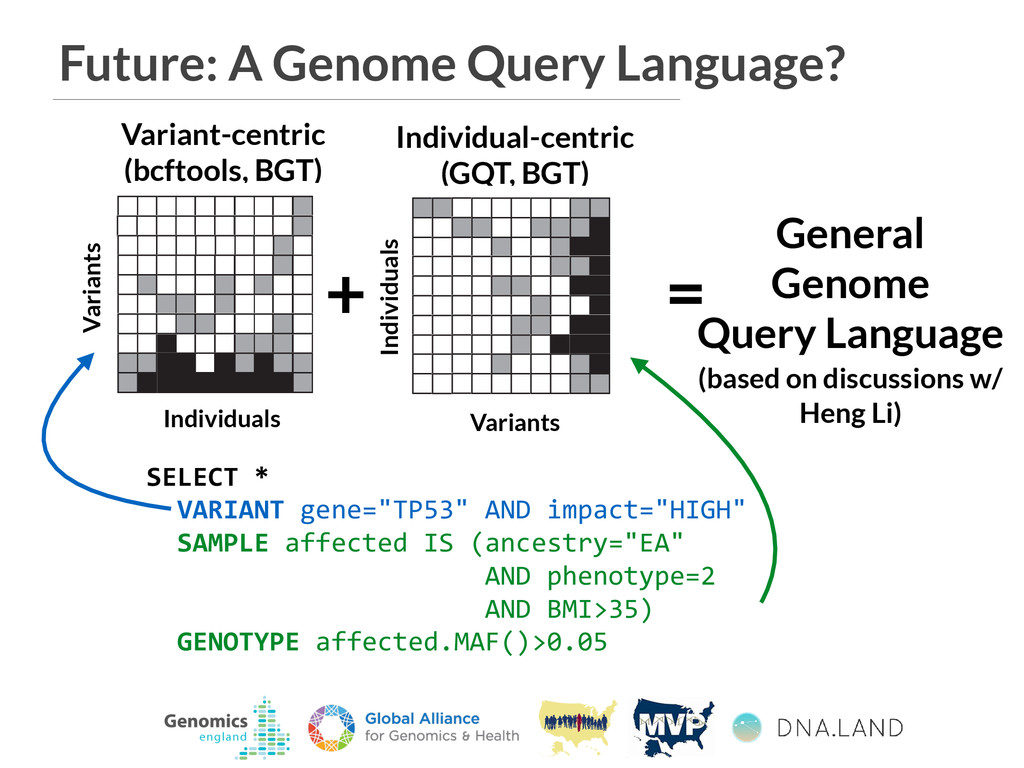
General Genome Query Language (based on discussions w/ Heng Li) VCF A B PED SQL database GQT index Individuals Variants 3 4 5 6 9 gqt convert ped gqt convert vcf D C Find variants that are common in cases and rare in controls. b gqt query study.gqt study.db -p "phenotype == 2" -g "maf() > 0.05" -p "phenotype == 1" -g "maf() < 0.05" gqt -p -g b VCF In F V In VCF A B PED SQL database GQT index Individuals Variants 3 4 5 6 9 gqt convert ped gqt convert vcf D C Find variants that are common in g Individual-centric (GQT, BGT) Individuals Variants Variants Individuals
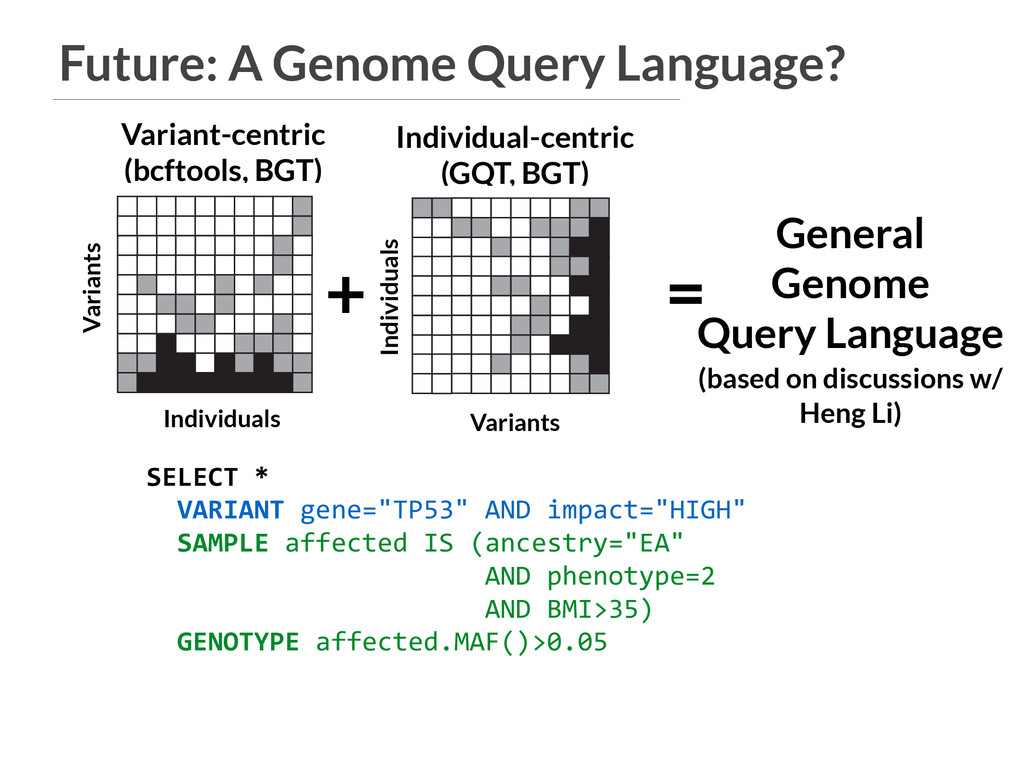
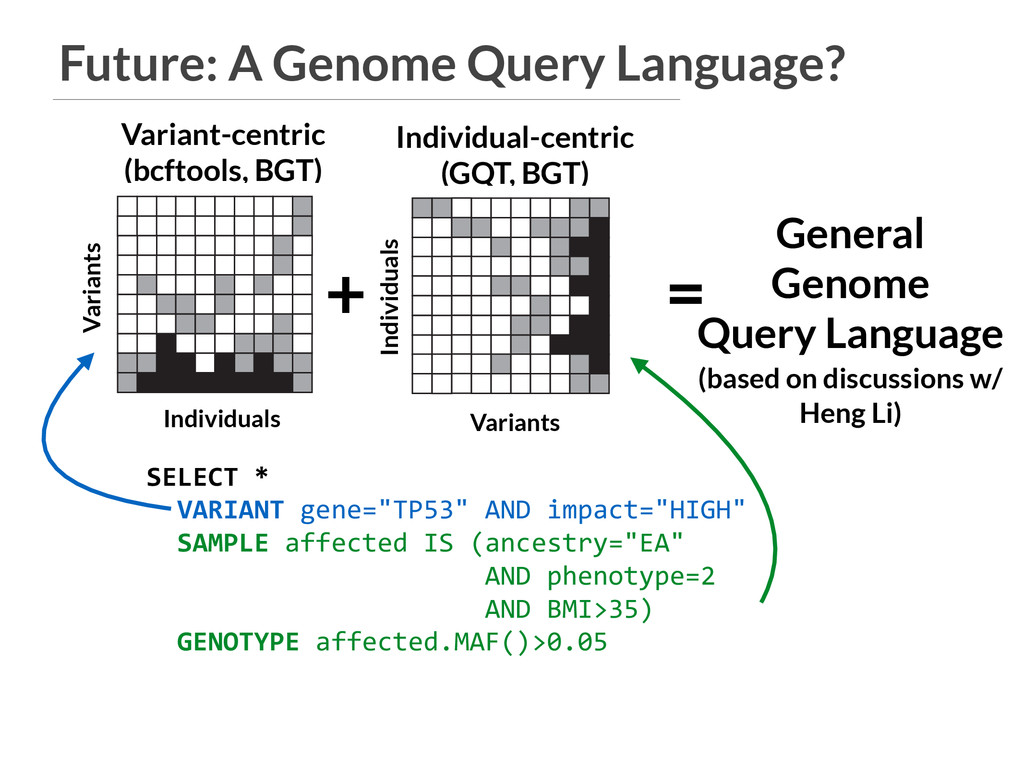
General Genome Query Language (based on discussions w/ Heng Li) VCF A B PED SQL database GQT index Individuals Variants 3 4 5 6 9 gqt convert ped gqt convert vcf D C Find variants that are common in cases and rare in controls. b gqt query study.gqt study.db -p "phenotype == 2" -g "maf() > 0.05" -p "phenotype == 1" -g "maf() < 0.05" gqt -p -g b VCF In F V In VCF A B PED SQL database GQT index Individuals Variants 3 4 5 6 9 gqt convert ped gqt convert vcf D C Find variants that are common in g Individual-centric (GQT, BGT) Individuals Variants Variants Individuals SELECT * VARIANT gene="TP53" AND impact="HIGH" SAMPLE affected IS (ancestry="EA" AND phenotype=2 AND BMI>35) GENOTYPE affected.MAF()>0.05
General Genome Query Language (based on discussions w/ Heng Li) VCF A B PED SQL database GQT index Individuals Variants 3 4 5 6 9 gqt convert ped gqt convert vcf D C Find variants that are common in cases and rare in controls. b gqt query study.gqt study.db -p "phenotype == 2" -g "maf() > 0.05" -p "phenotype == 1" -g "maf() < 0.05" gqt -p -g b VCF In F V In VCF A B PED SQL database GQT index Individuals Variants 3 4 5 6 9 gqt convert ped gqt convert vcf D C Find variants that are common in g Individual-centric (GQT, BGT) Individuals Variants Variants Individuals SELECT * VARIANT gene="TP53" AND impact="HIGH" SAMPLE affected IS (ancestry="EA" AND phenotype=2 AND BMI>35) GENOTYPE affected.MAF()>0.05
General Genome Query Language (based on discussions w/ Heng Li) VCF A B PED SQL database GQT index Individuals Variants 3 4 5 6 9 gqt convert ped gqt convert vcf D C Find variants that are common in cases and rare in controls. b gqt query study.gqt study.db -p "phenotype == 2" -g "maf() > 0.05" -p "phenotype == 1" -g "maf() < 0.05" gqt -p -g b VCF In F V In VCF A B PED SQL database GQT index Individuals Variants 3 4 5 6 9 gqt convert ped gqt convert vcf D C Find variants that are common in g Individual-centric (GQT, BGT) Individuals Variants Variants Individuals SELECT * VARIANT gene="TP53" AND impact="HIGH" SAMPLE affected IS (ancestry="EA" AND phenotype=2 AND BMI>35) GENOTYPE affected.MAF()>0.05
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[[annotation]] file=“ExAC.v3.vcf” fields=[“AF”, “AC_Het”] names=[“exac_aaf”, “exac_num_het”]](https://files.speakerdeck.com/presentations/ca375488cf434cf79151191d087e658a/slide_5.jpg){kind=link}
![[[annotation]] file=“ExAC.v3.vcf” fields=[“AF”, “AC_Het”] names=[“exac_aaf”, “exac_num_het”]](https://files.speakerdeck.com/presentations/ca375488cf434cf79151191d087e658a/slide_6.jpg){kind=link}
![[[annotation]] file=“ExAC.v3.vcf” fields=[“AF”, “AC_Het”] names=[“exac_aaf”, “exac_num_het”]](https://files.speakerdeck.com/presentations/ca375488cf434cf79151191d087e658a/slide_7.jpg){kind=link}
![[[annotation]] file=“ExAC.v3.vcf” fields=[“AF”, “AC_Het”] names=[“exac_aaf”, “exac_num_het”]](https://files.speakerdeck.com/presentations/ca375488cf434cf79151191d087e658a/slide_8.jpg){kind=link}
![[[annotation]] file=“ExAC.v3.vcf” fields=[“AF”, “AC_Het”] names=[“exac_aaf”, “exac_num_het”]](https://files.speakerdeck.com/presentations/ca375488cf434cf79151191d087e658a/slide_9.jpg){kind=link}
![[[annotation]] file=“ExAC.v3.vcf” fields=[“AF”, “AC_Het”] names=[“exac_aaf”, “exac_num_het”]](https://files.speakerdeck.com/presentations/ca375488cf434cf79151191d087e658a/slide_10.jpg){kind=link}
{kind=link}
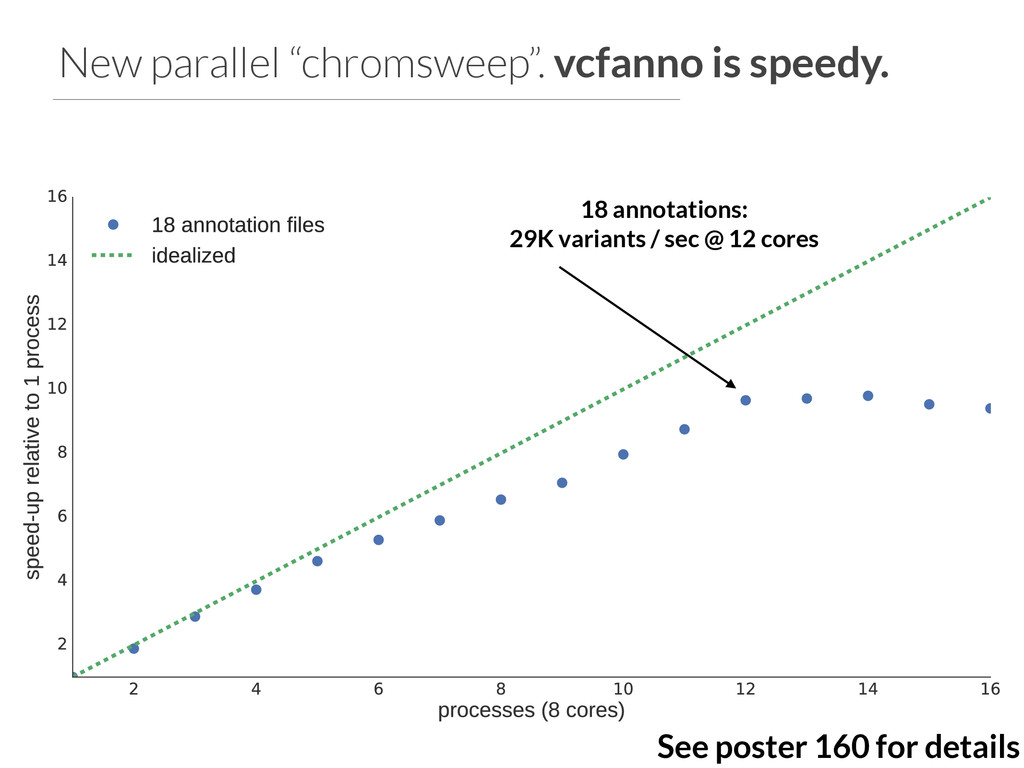{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
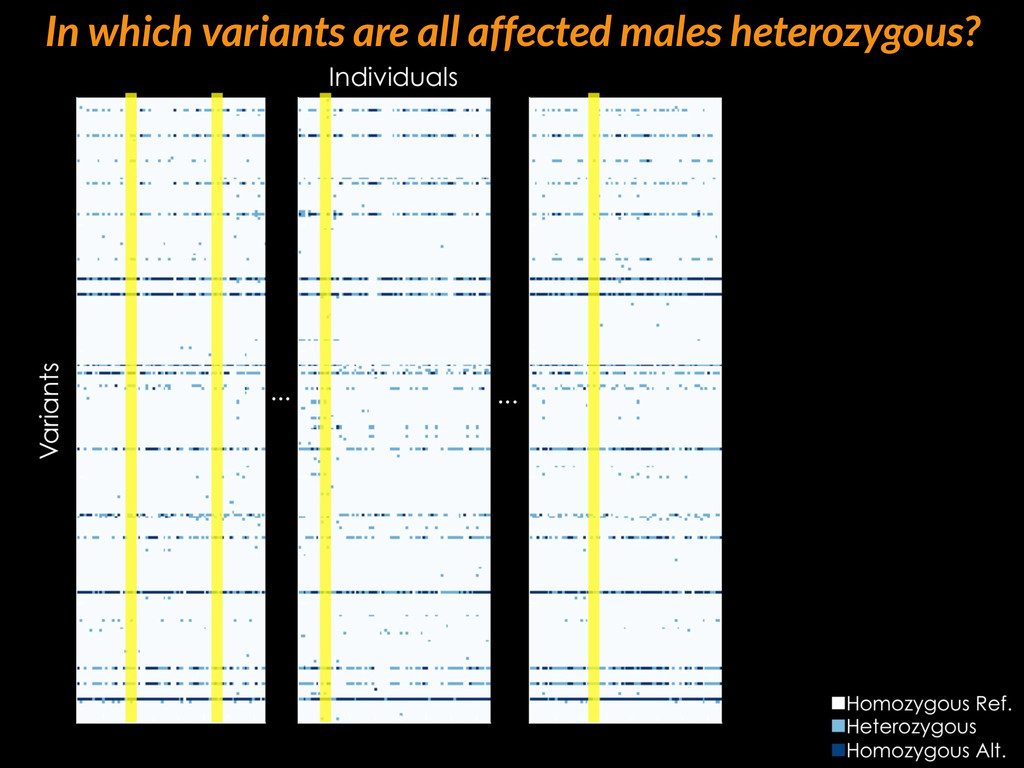{kind=link}
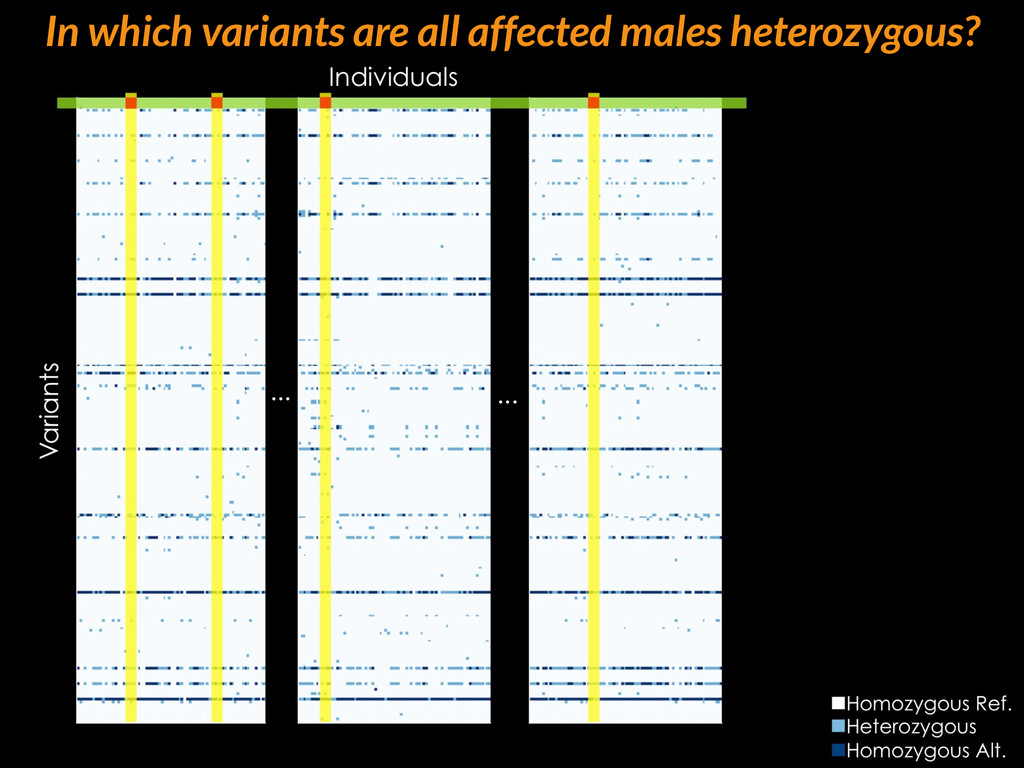{kind=link}
{kind=link}
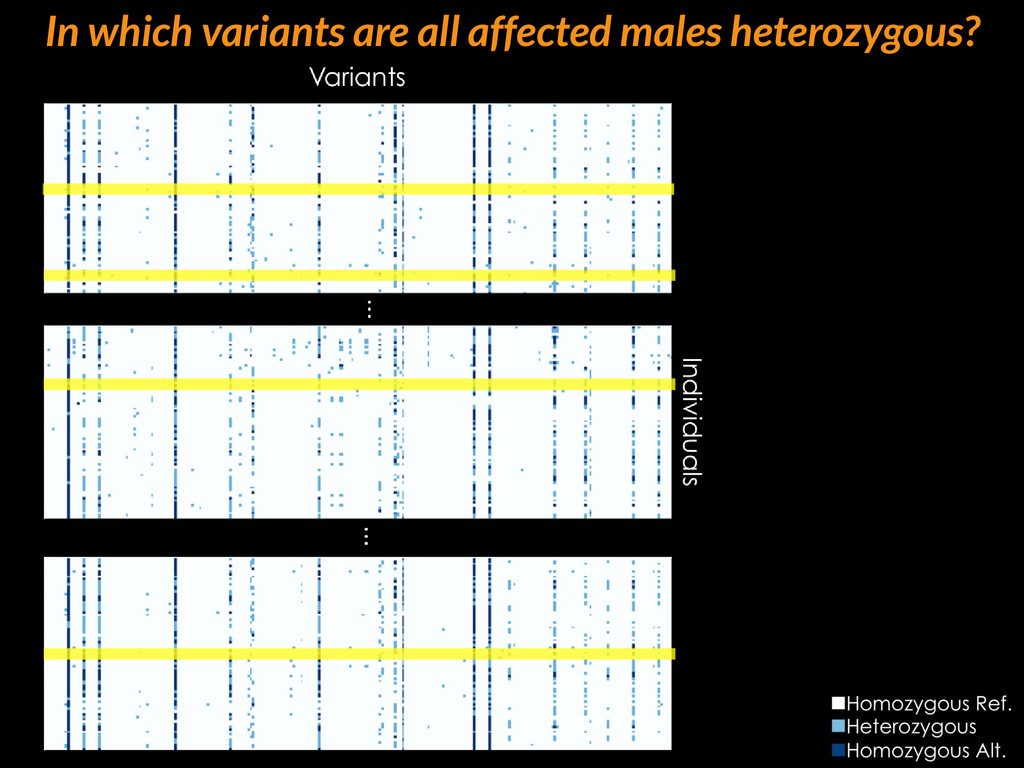{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}