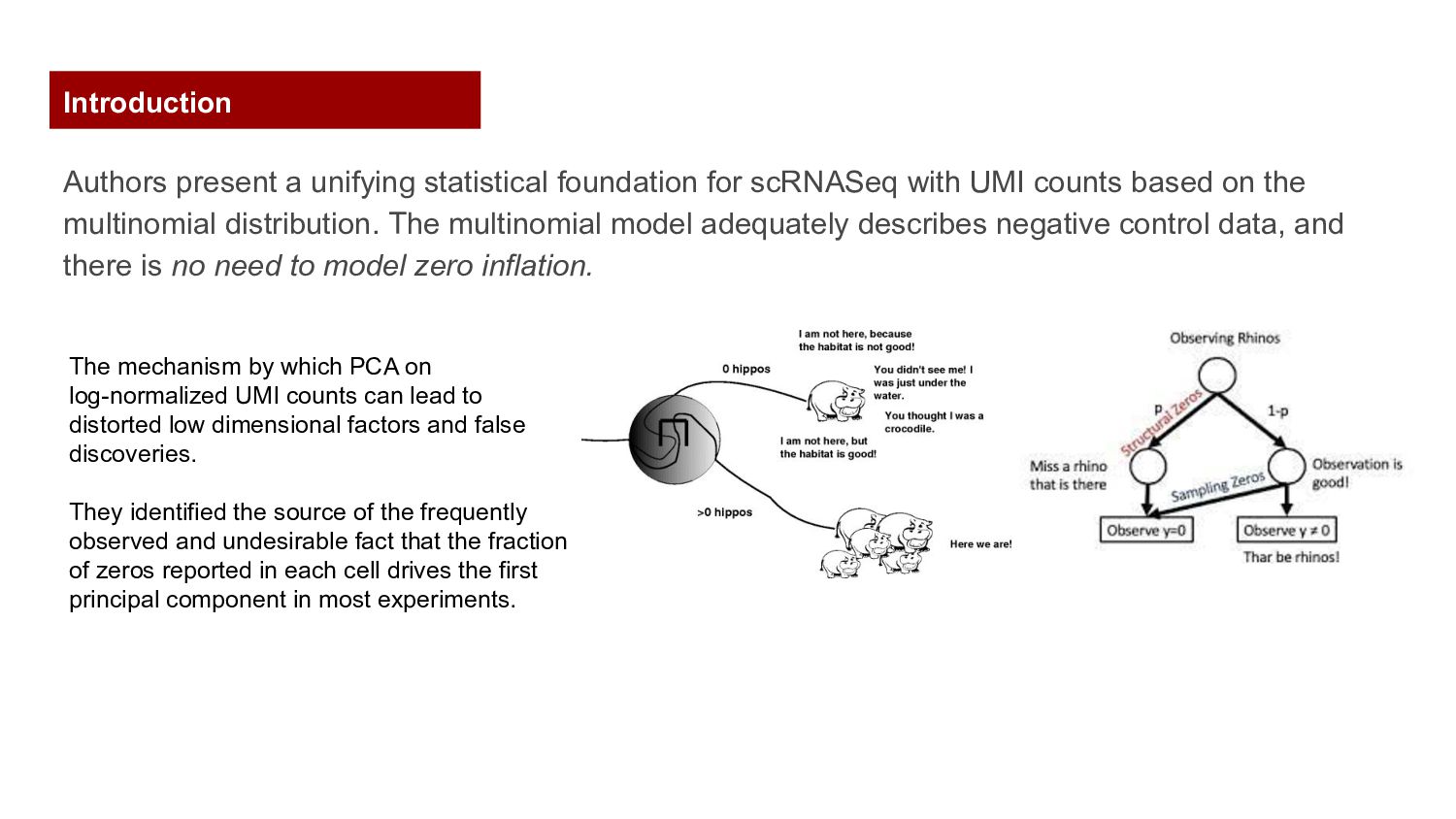
counts based on the multinomial distribution. The multinomial model adequately describes negative control data, and there is no need to model zero inflation. The mechanism by which PCA on log-normalized UMI counts can lead to distorted low dimensional factors and false discoveries. They identified the source of the frequently observed and undesirable fact that the fraction of zeros reported in each cell drives the first principal component in most experiments. Introduction
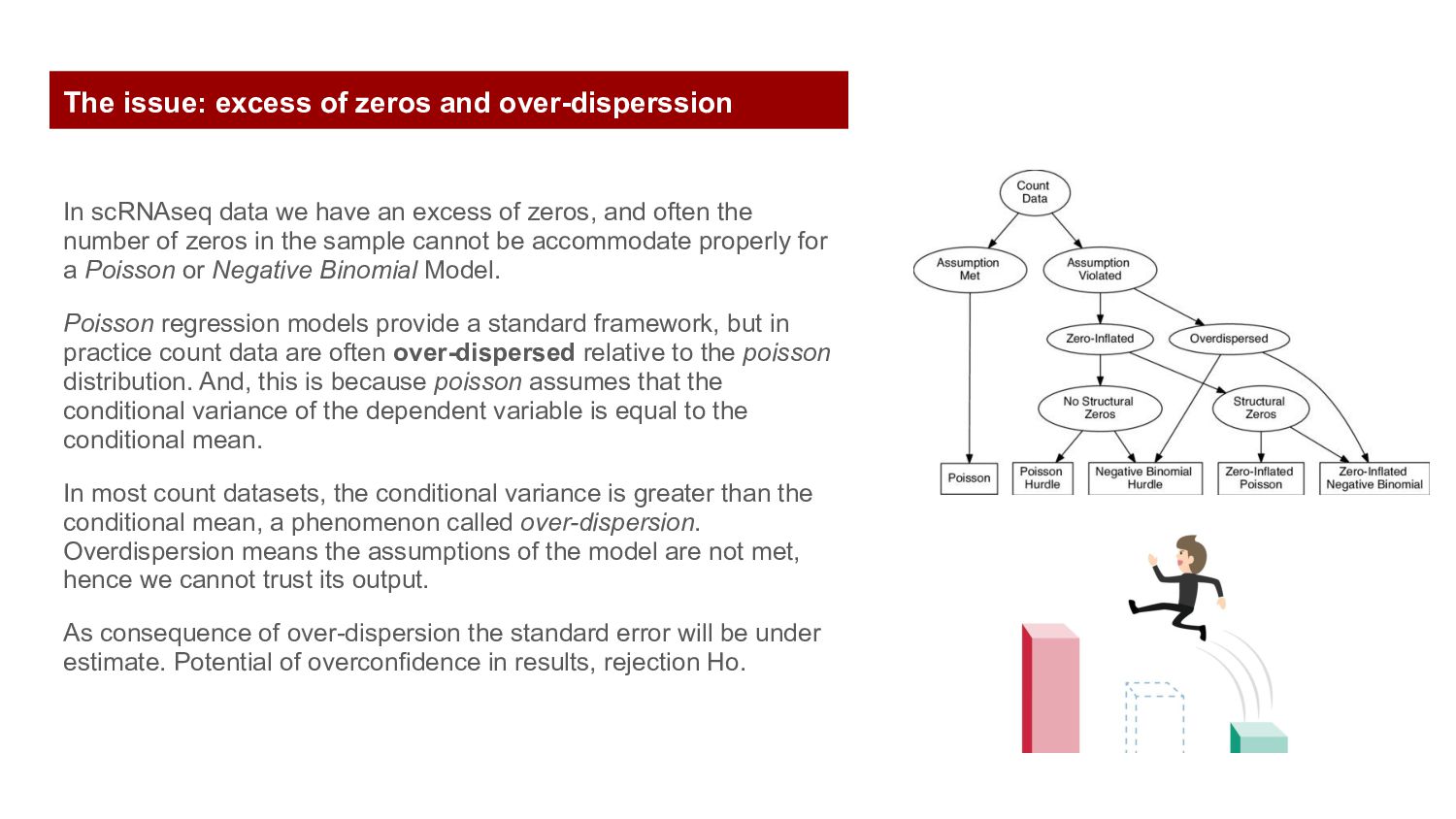
often the number of zeros in the sample cannot be accommodate properly for a Poisson or Negative Binomial Model. Poisson regression models provide a standard framework, but in practice count data are often over-dispersed relative to the poisson distribution. And, this is because poisson assumes that the conditional variance of the dependent variable is equal to the conditional mean. In most count datasets, the conditional variance is greater than the conditional mean, a phenomenon called over-dispersion. Overdispersion means the assumptions of the model are not met, hence we cannot trust its output. As consequence of over-dispersion the standard error will be under estimate. Potential of overconfidence in results, rejection Ho. The issue: excess of zeros and over-disperssion
to describe the sampling distribution of genes with high counts and the negative binomial model is more appropriate. The benefits of avoiding normalization by instead directly modeling raw counts have been demonstrated in the context of differential expression. Where normalization is unavoidable, authors propose the use of approximate multinomial deviance residuals. The issue: excess of zeros and over-disperssion
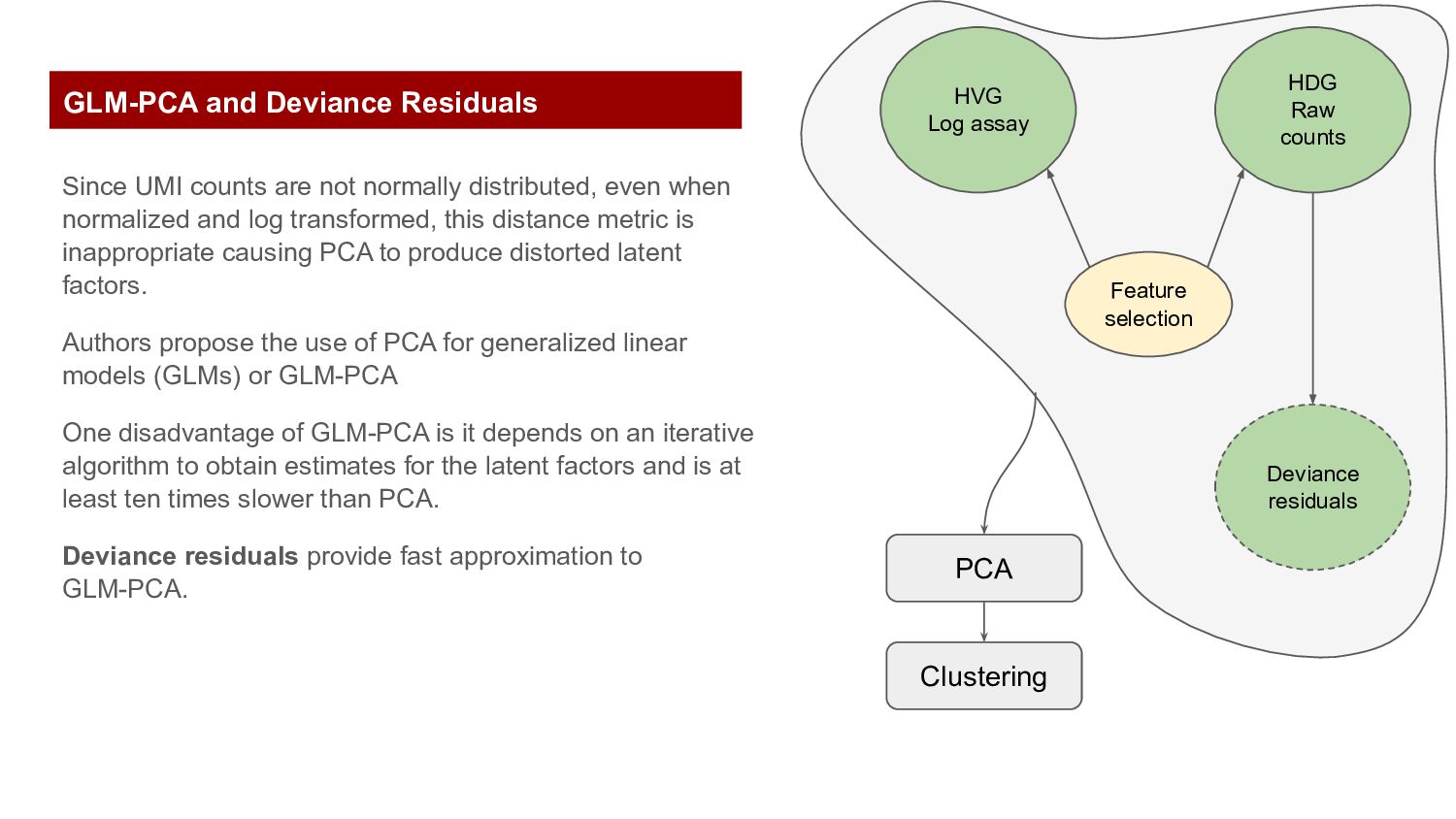
and log transformed, this distance metric is inappropriate causing PCA to produce distorted latent factors. Authors propose the use of PCA for generalized linear models (GLMs) or GLM-PCA One disadvantage of GLM-PCA is it depends on an iterative algorithm to obtain estimates for the latent factors and is at least ten times slower than PCA. Deviance residuals provide fast approximation to GLM-PCA. Feature selection HVG Log assay HDG Raw counts Deviance residuals PCA Clustering GLM-PCA and Deviance Residuals
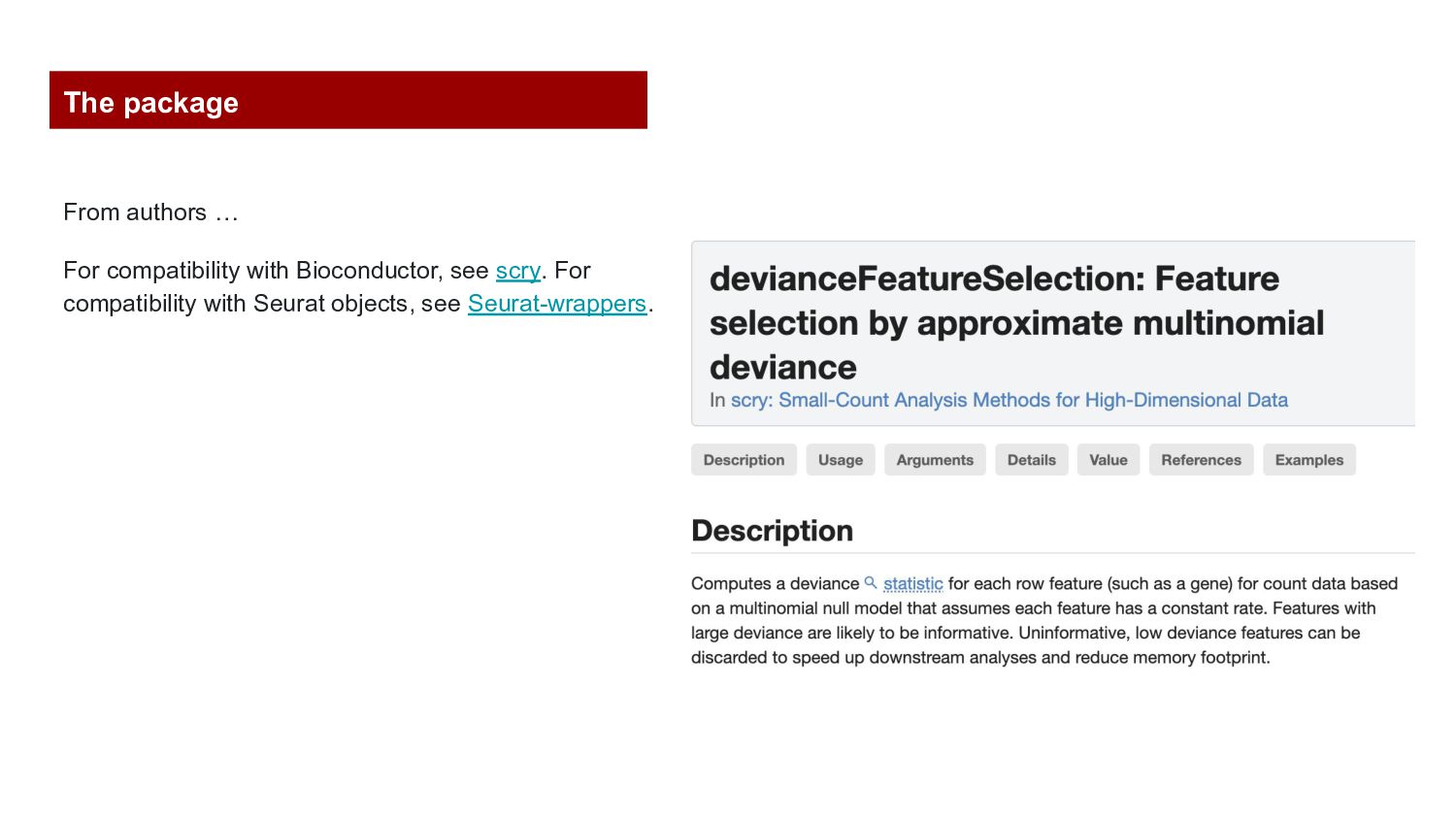
variance of the log-expression profiles for each gene. 2. Define a set of highly variable genes, based on variance modelling statistics a. getTopHVGs: PC_fdr1, PC_fdr5, PC_p1, PC_p2, PC_p5) 3. runPCA: PC_fdr1 1. Compute GLM-PCA: Computes a deviance statistic for each gene for count data based on a multinomial null model that assumes each feature has a constant rate. Binomial_deviance in counts assay Thresholds: hdgs.hb.2000, hdgs.hb.5000, hdgs.hb.10000 2. runPCA: binomial_deviance_residuals in the selected thresholds.
objects, see Seurat-wrappers. Alternative implementations GLM-PCA has been around for awhile and we have not been able to dedicate as much time to its maintenance and ongoing improvement as we would like. Fortunately, there are numerous alternative implementations that improve on our basic idea. Here are some packages to check out. • fastglmpca. Preprint: Weine, Carbonetto, & Stephens (2024). • scGBM. Preprint: Nicol & Miller 2023. • NewWave. Publication: Agostinis et al 2022. • LDVAE. Publication: Svensson et al 2020. The package
implementations of count-based feature selection and dimension reduction algorithms. These methods can be used to facilitate unsupervised analysis of any high-dimensional data such as single-cell RNA-seq. Basic Workflow: vignettes/scry.Rmd More wide explanation in action: Modeling single cell RNAseq data with multinomial distribution Zero-inflated models: zero-inflated-models
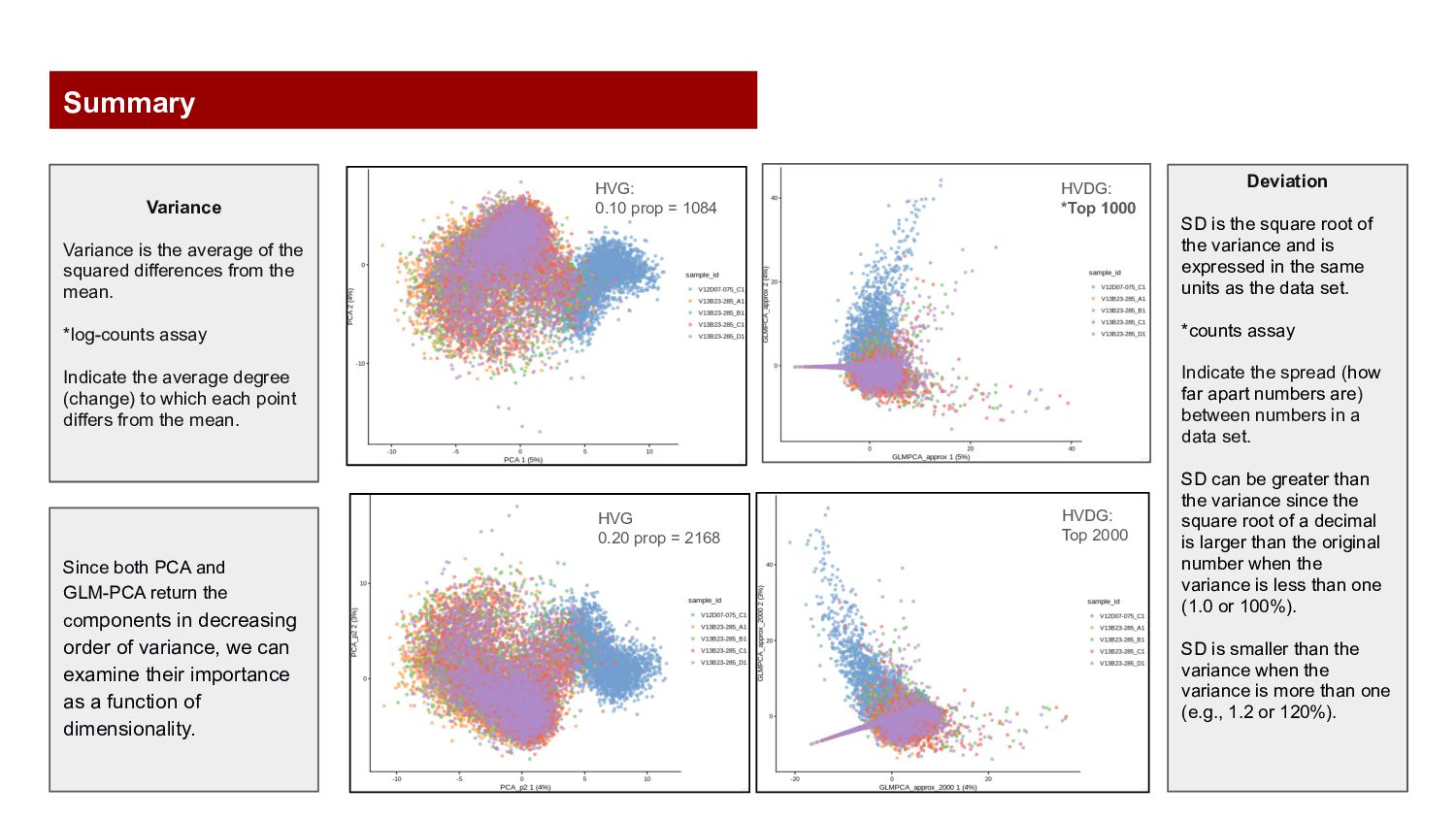
prop = 2168 HVDG: *Top 1000 Deviation SD is the square root of the variance and is expressed in the same units as the data set. *counts assay Indicate the spread (how far apart numbers are) between numbers in a data set. SD can be greater than the variance since the square root of a decimal is larger than the original number when the variance is less than one (1.0 or 100%). SD is smaller than the variance when the variance is more than one (e.g., 1.2 or 120%). Variance Variance is the average of the squared differences from the mean. *log-counts assay Indicate the average degree (change) to which each point differs from the mean. Since both PCA and GLM-PCA return the components in decreasing order of variance, we can examine their importance as a function of dimensionality. Summary
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}