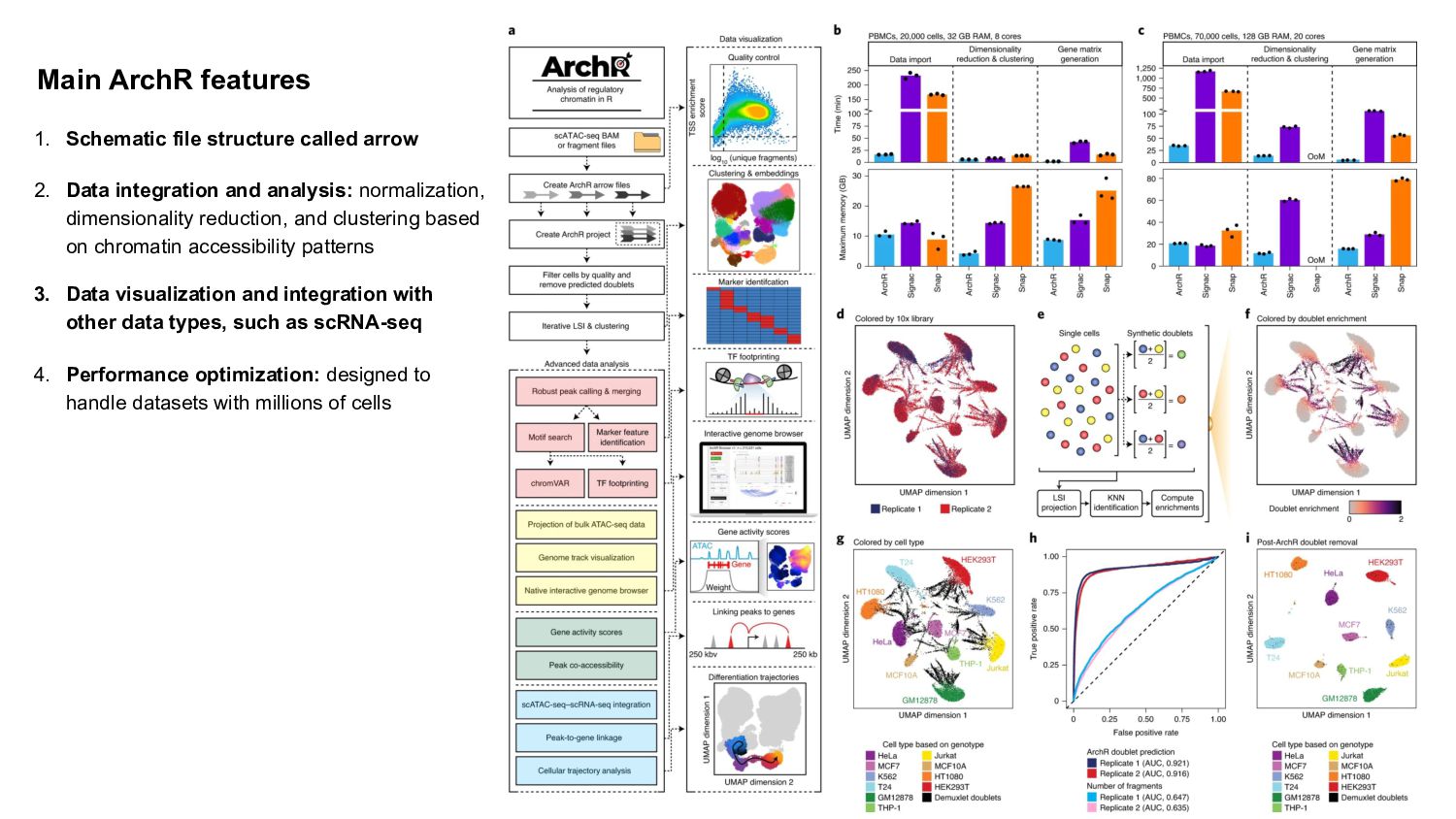
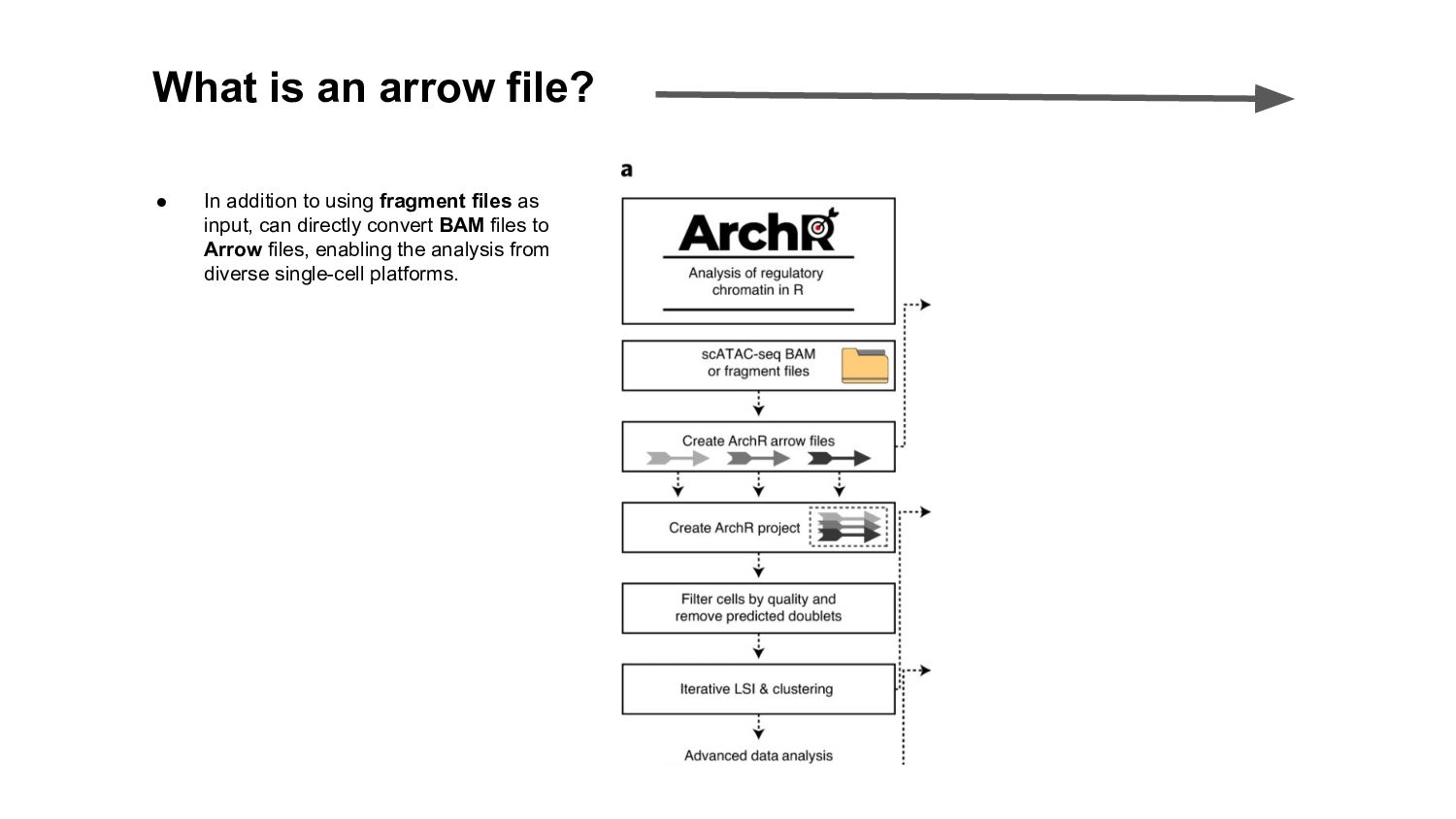
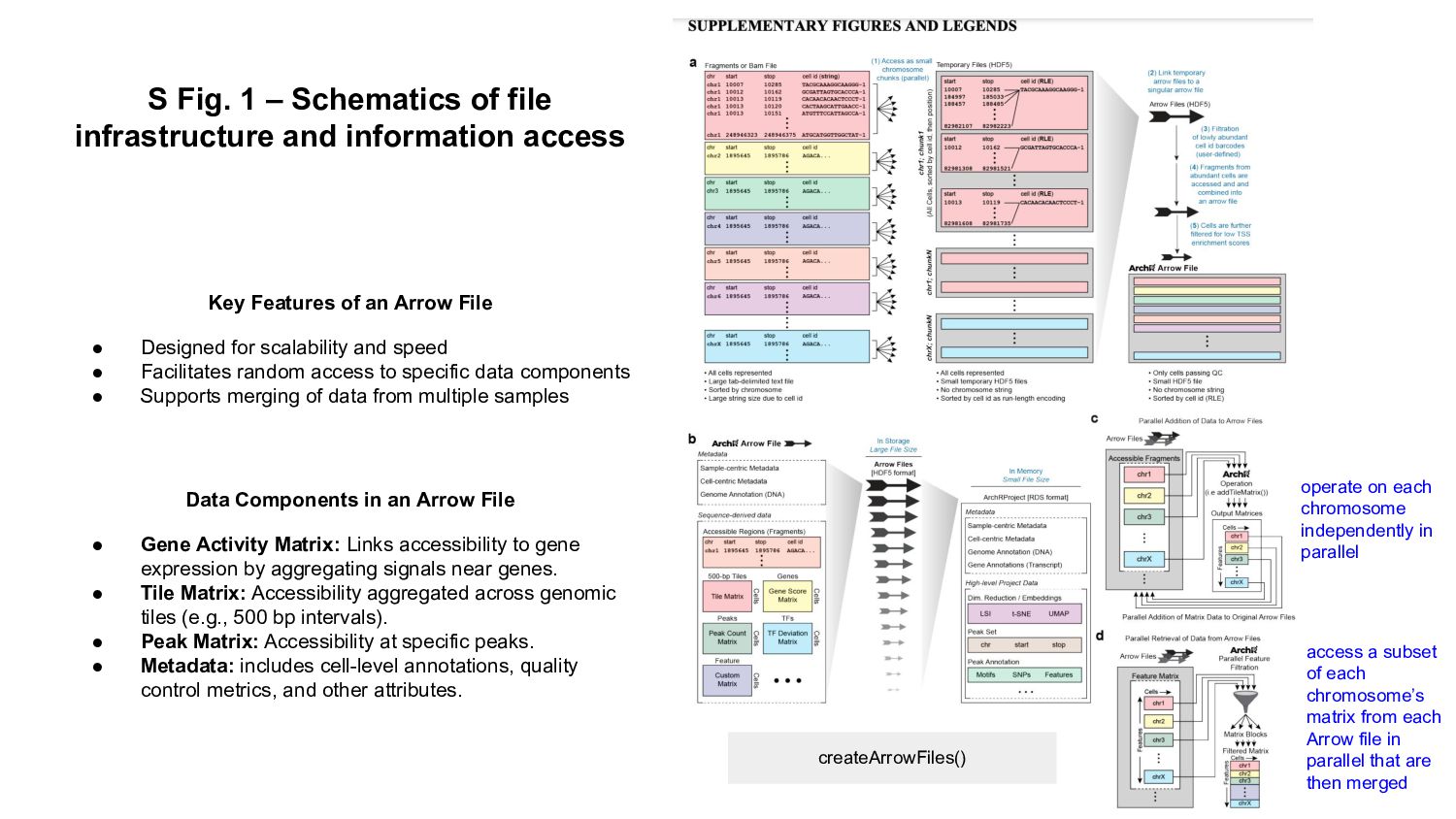
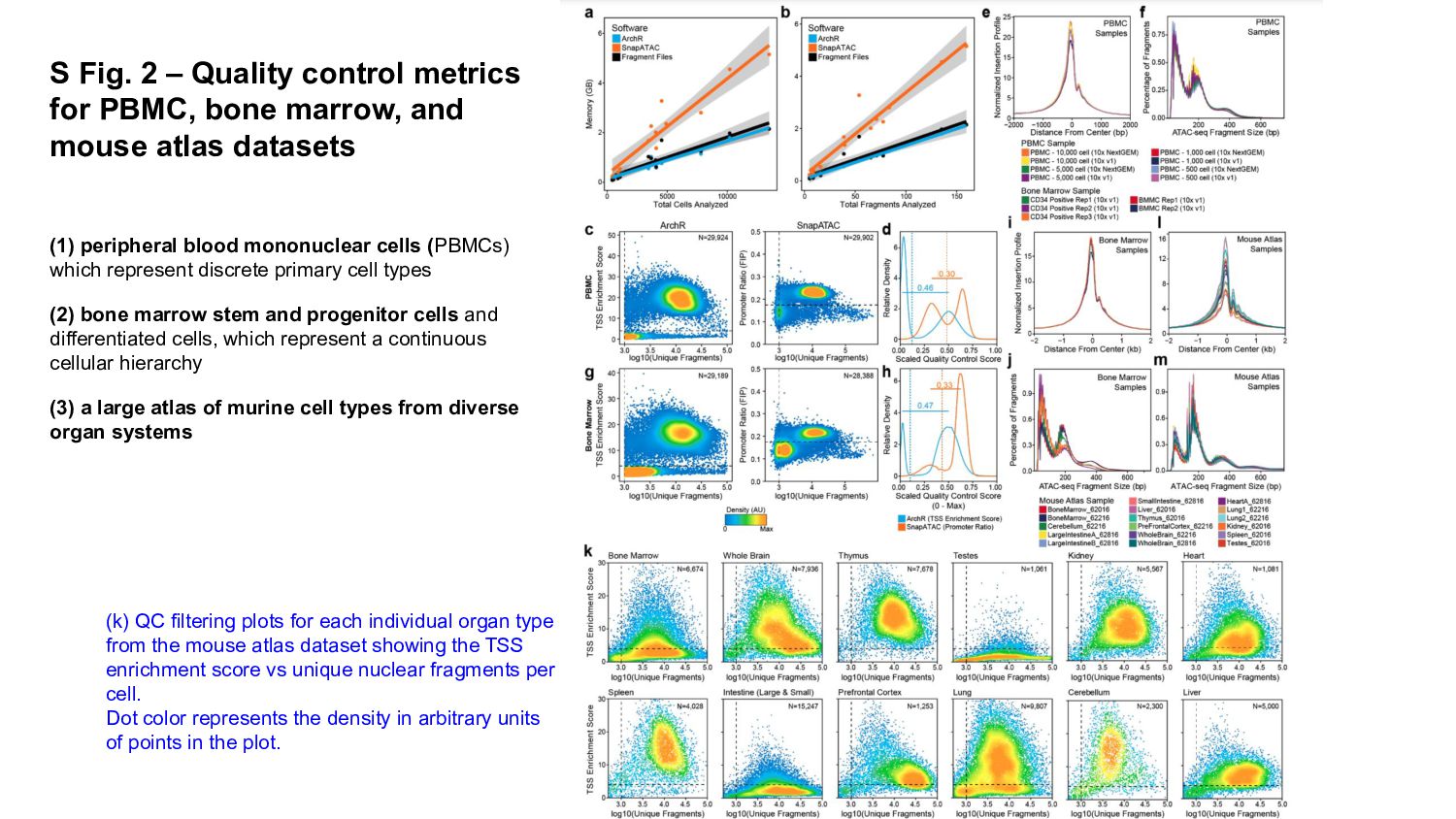
In this presentation I review ArchR software, a comprehensive and scalable R package designed for integrative single-cell chromatin accessibility analysis, excelling in handling large datasets and seamlessly integrating scATAC-seq with single-cell RNA-seq for multi-modal studies.
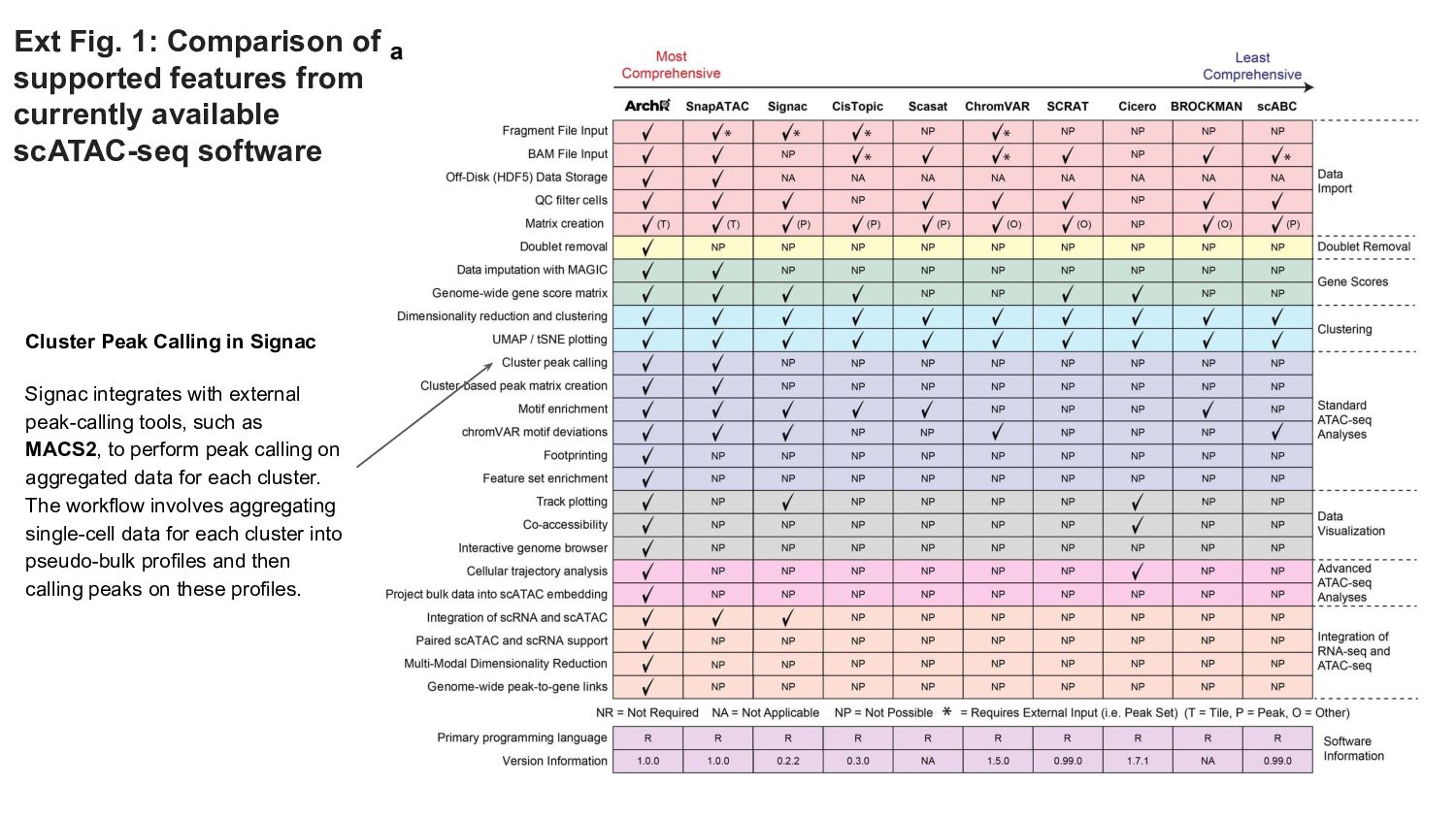
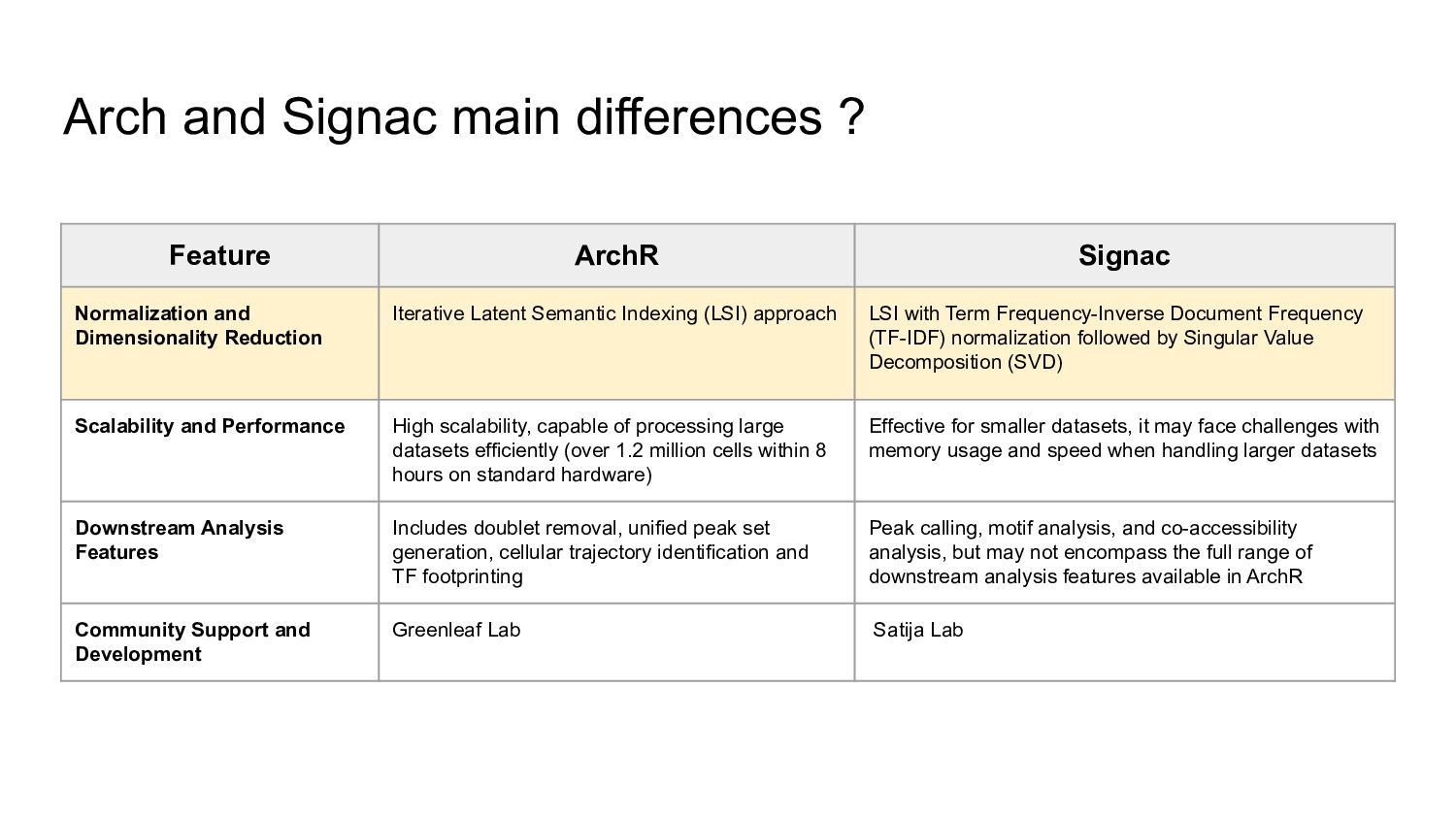
ArchR offers a user-friendly workflow with advanced features such as trajectory analysis, pseudo-bulk profiling, and high-quality visualizations. Compared to other tools, Signac, which extends Seurat for small to medium datasets, provides a unified RNA + ATAC workflow too, but is less efficient for large-scale analyses, while snapATAC specializes in large-scale scATAC-seq data with efficient cell barcoding but lacks advanced visualization and integration capabilities. Making the ideal choice for researchers needing both scalability and extensive multi-modal functionalities could challenging, tried this content to gain a comprehensive gist.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
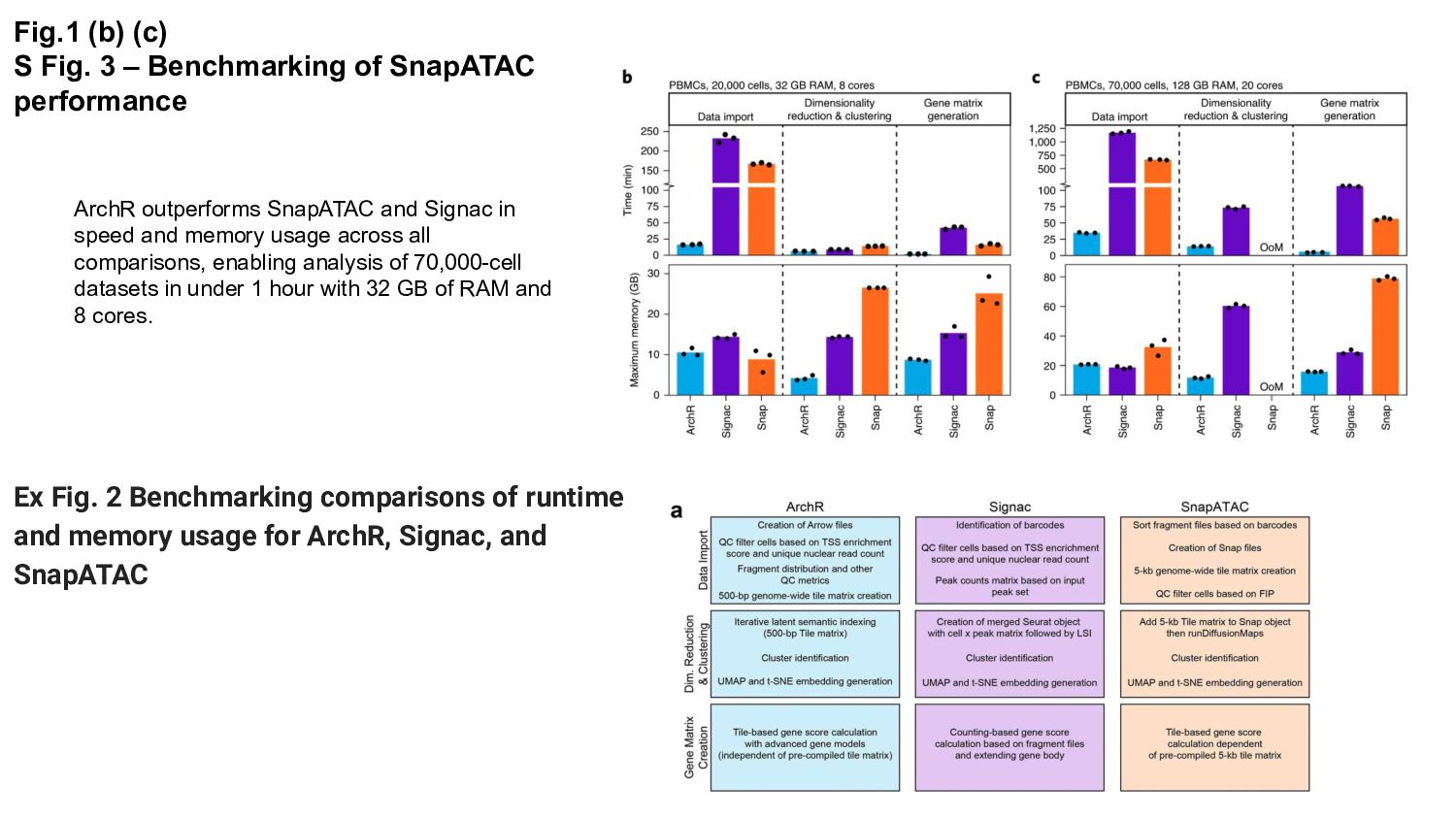{kind=link}
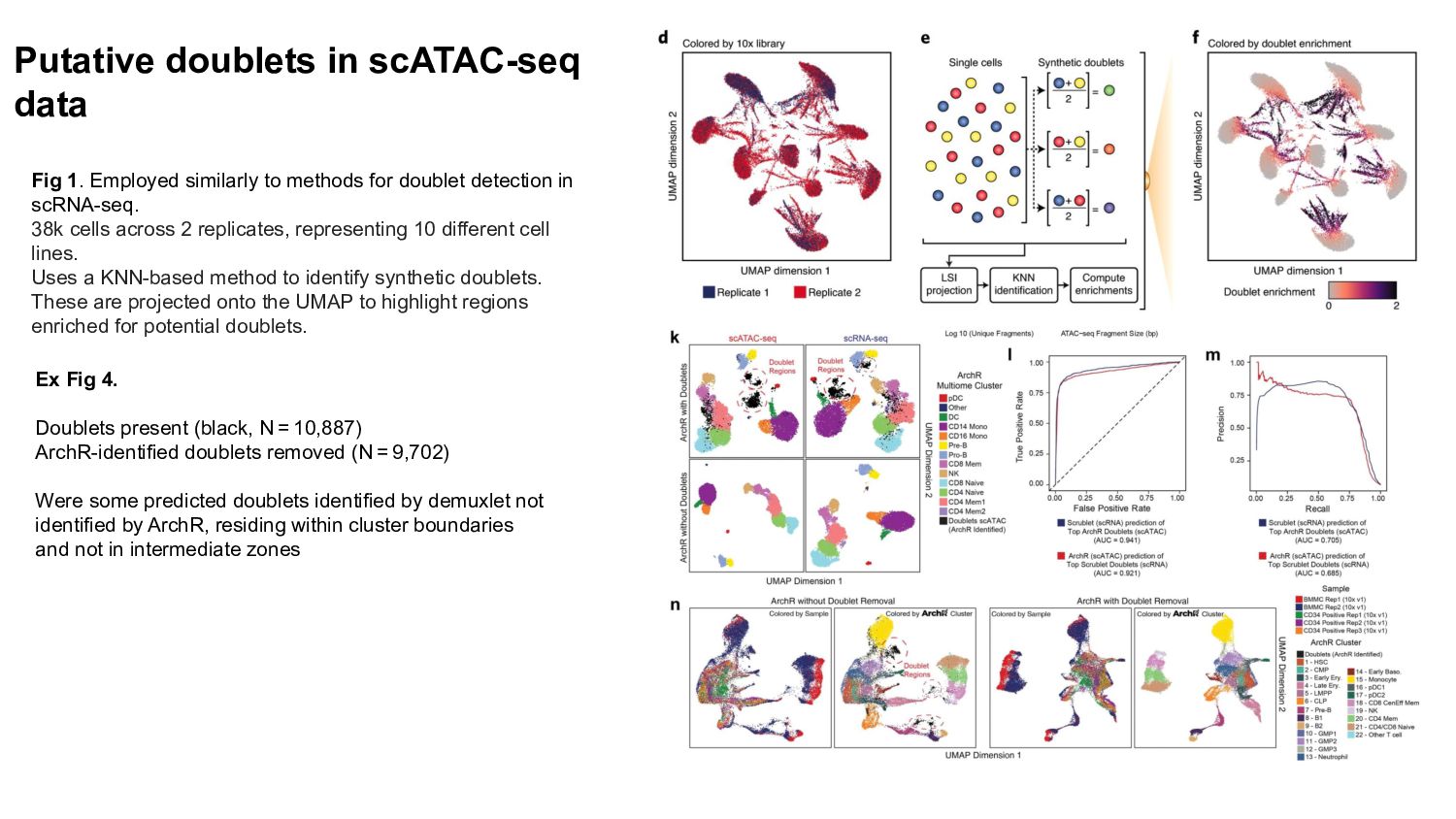{kind=link}
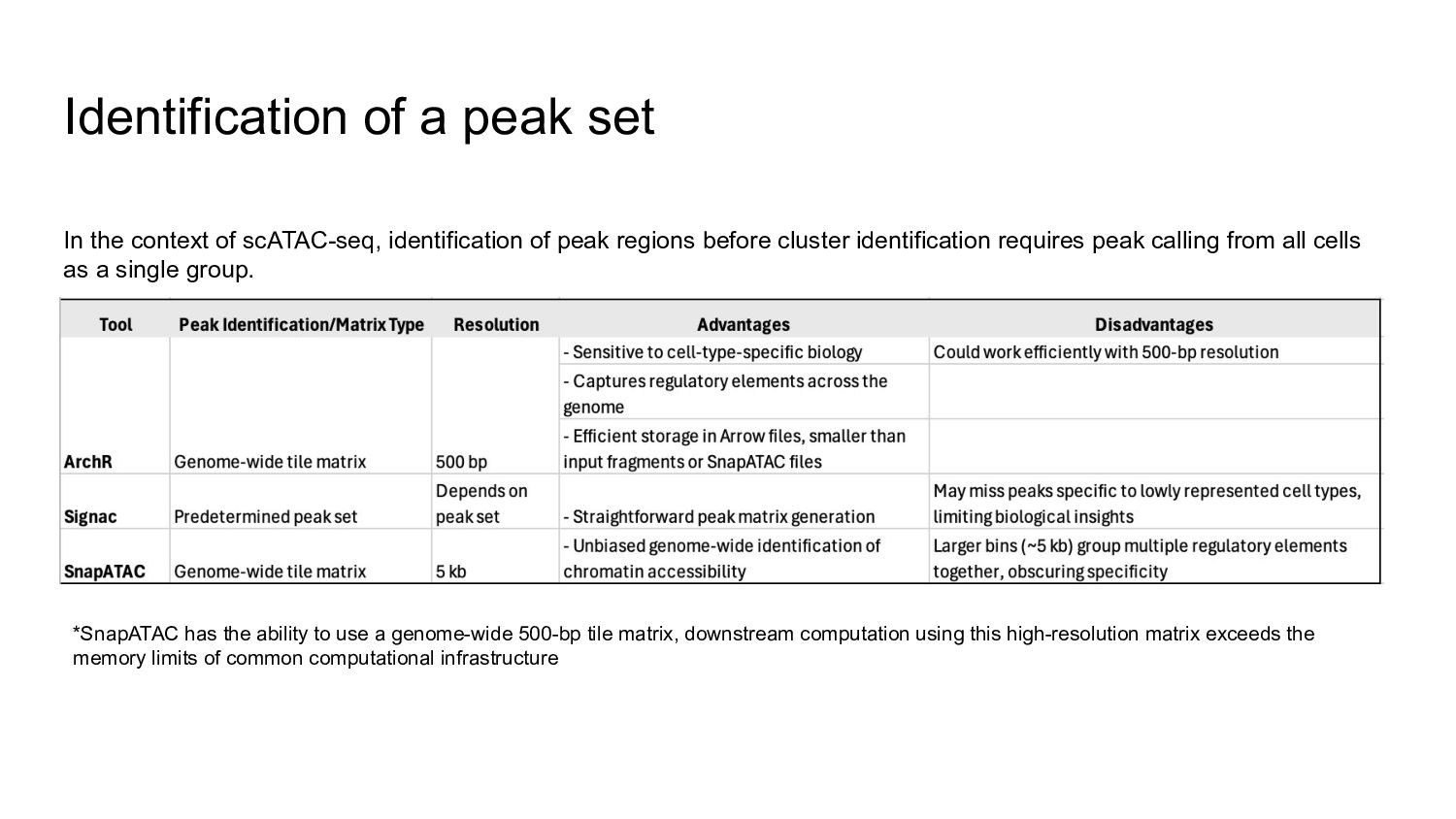{kind=link}
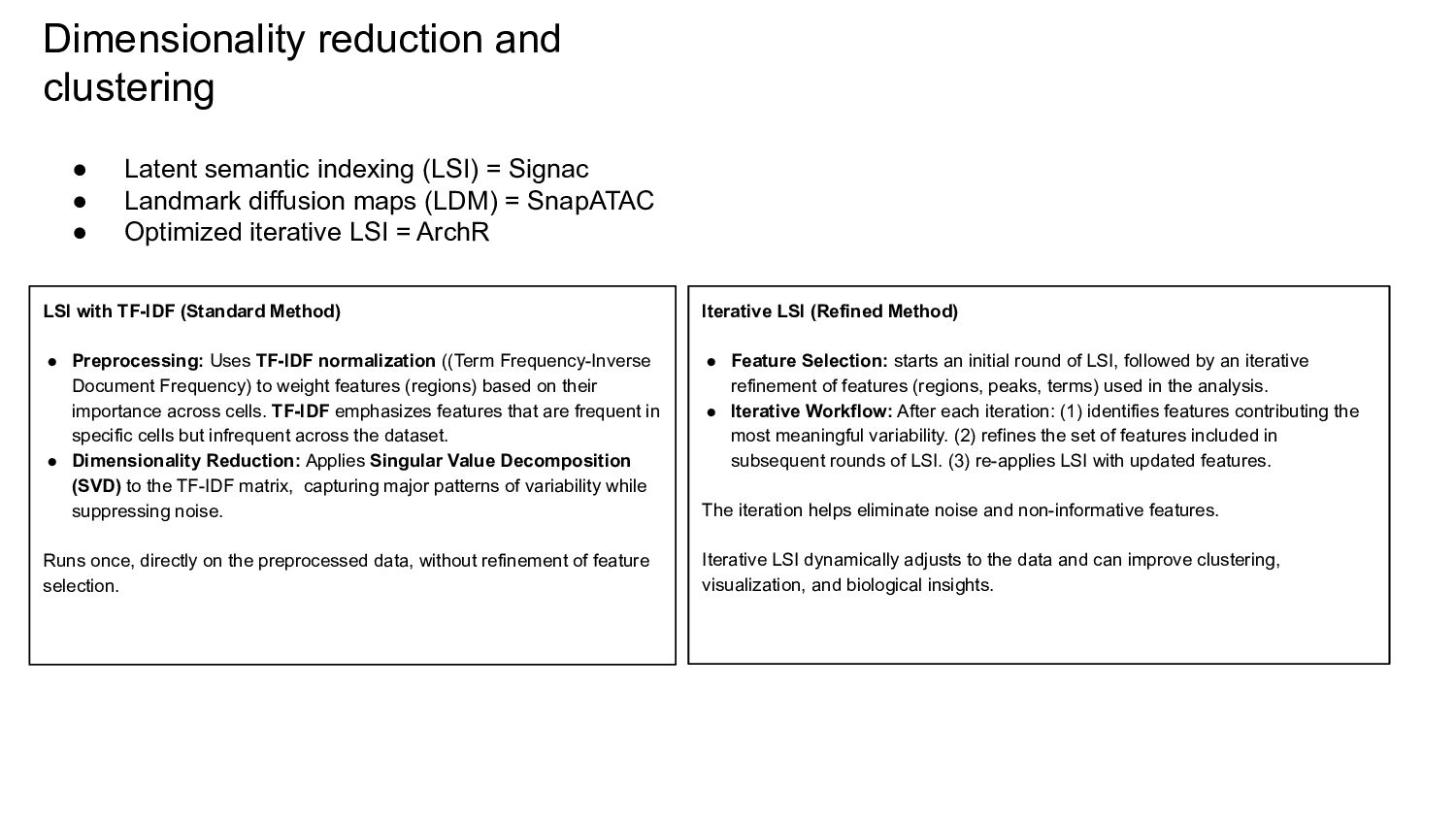{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}