Presenter:Cynthia Soto
Lieber Nov 6th, 2024
Human brain development spans from early embryogenesis through young adulthood, involving complex molecular changes as neurons differentiate, migrate, and form networks.
Disruptions to these processes by environmental or genetic factors may lead to neuropsychiatric disorders, necessitating a comprehensive view of developmental risk across all stages.
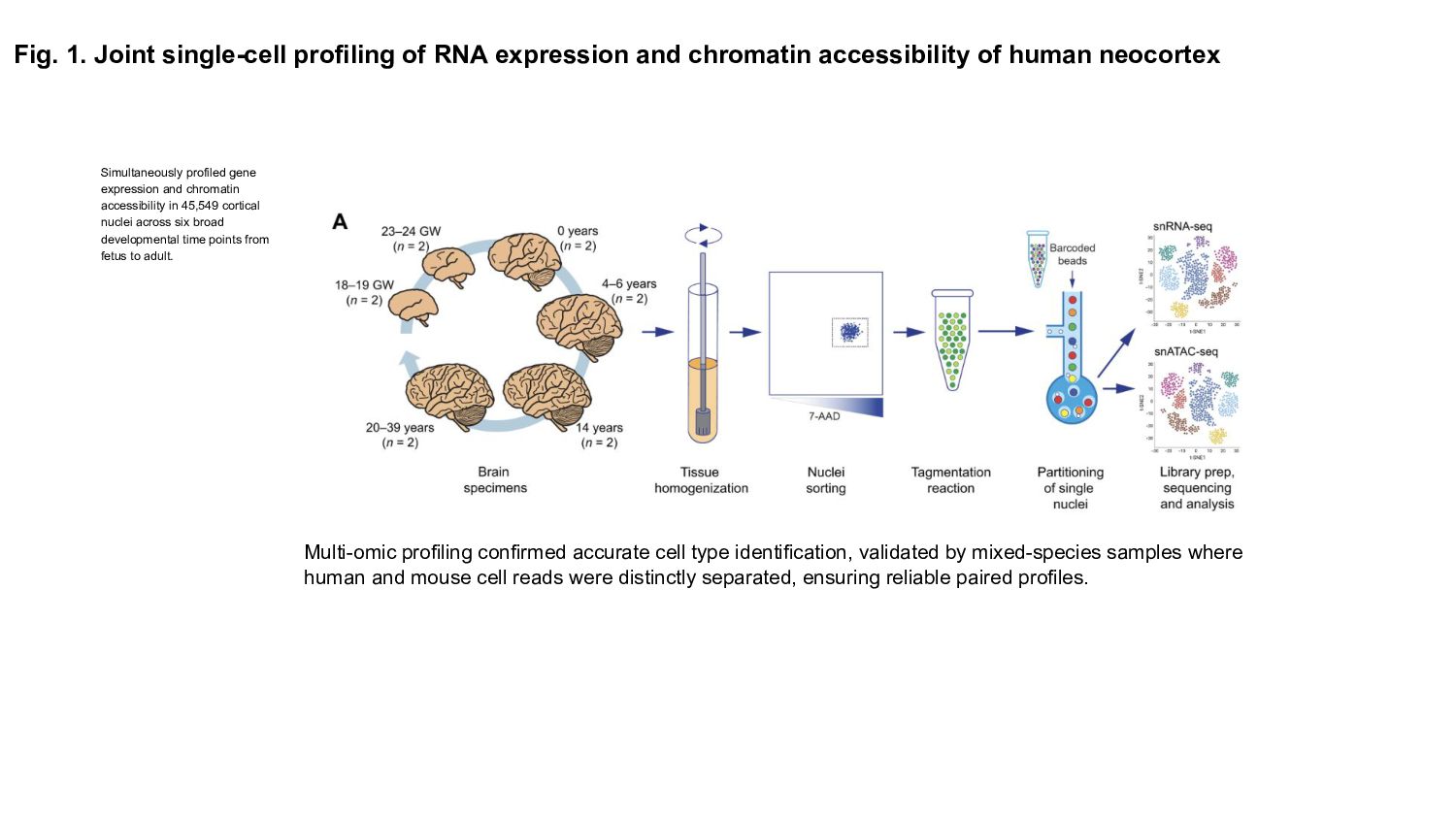
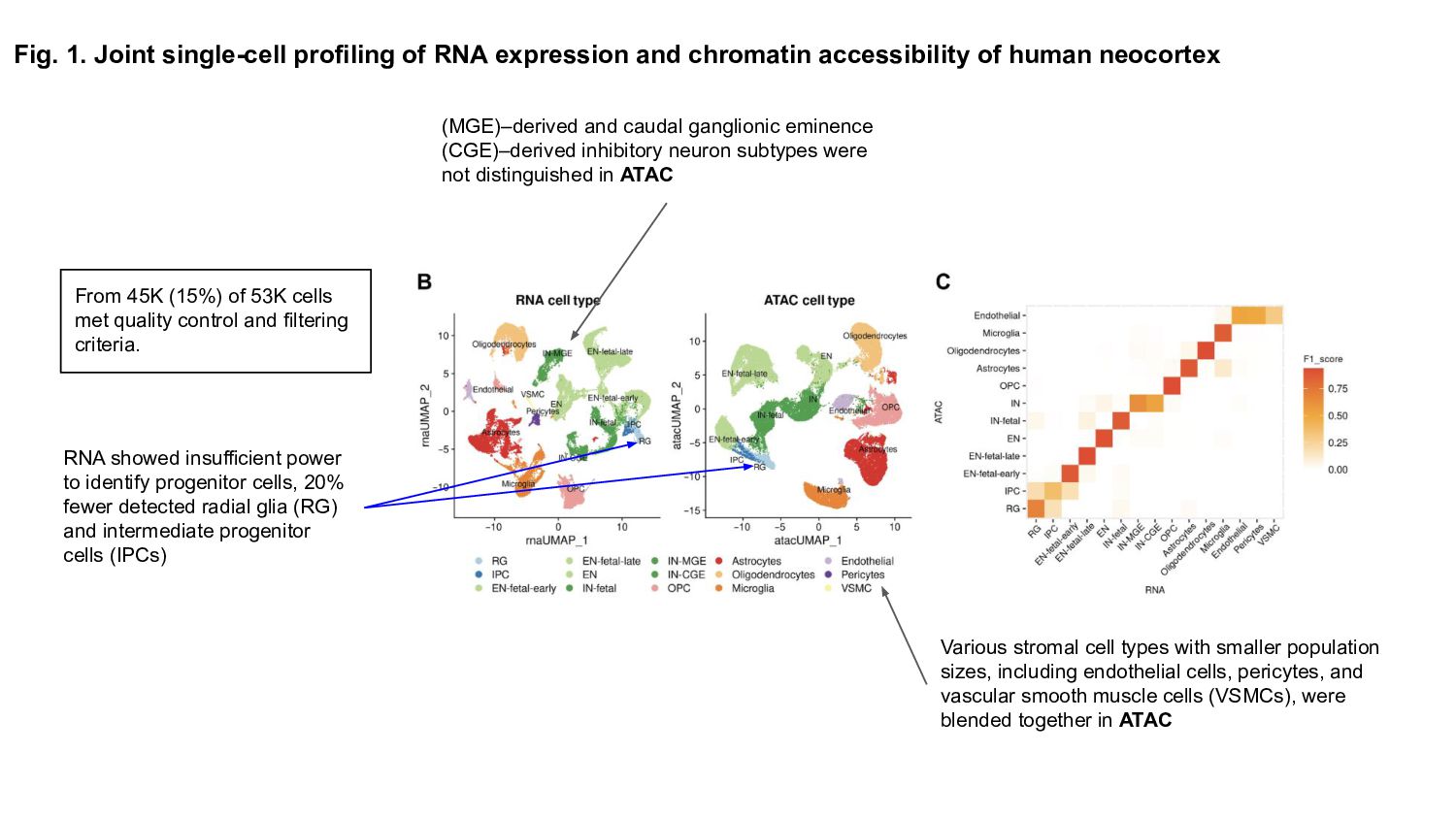
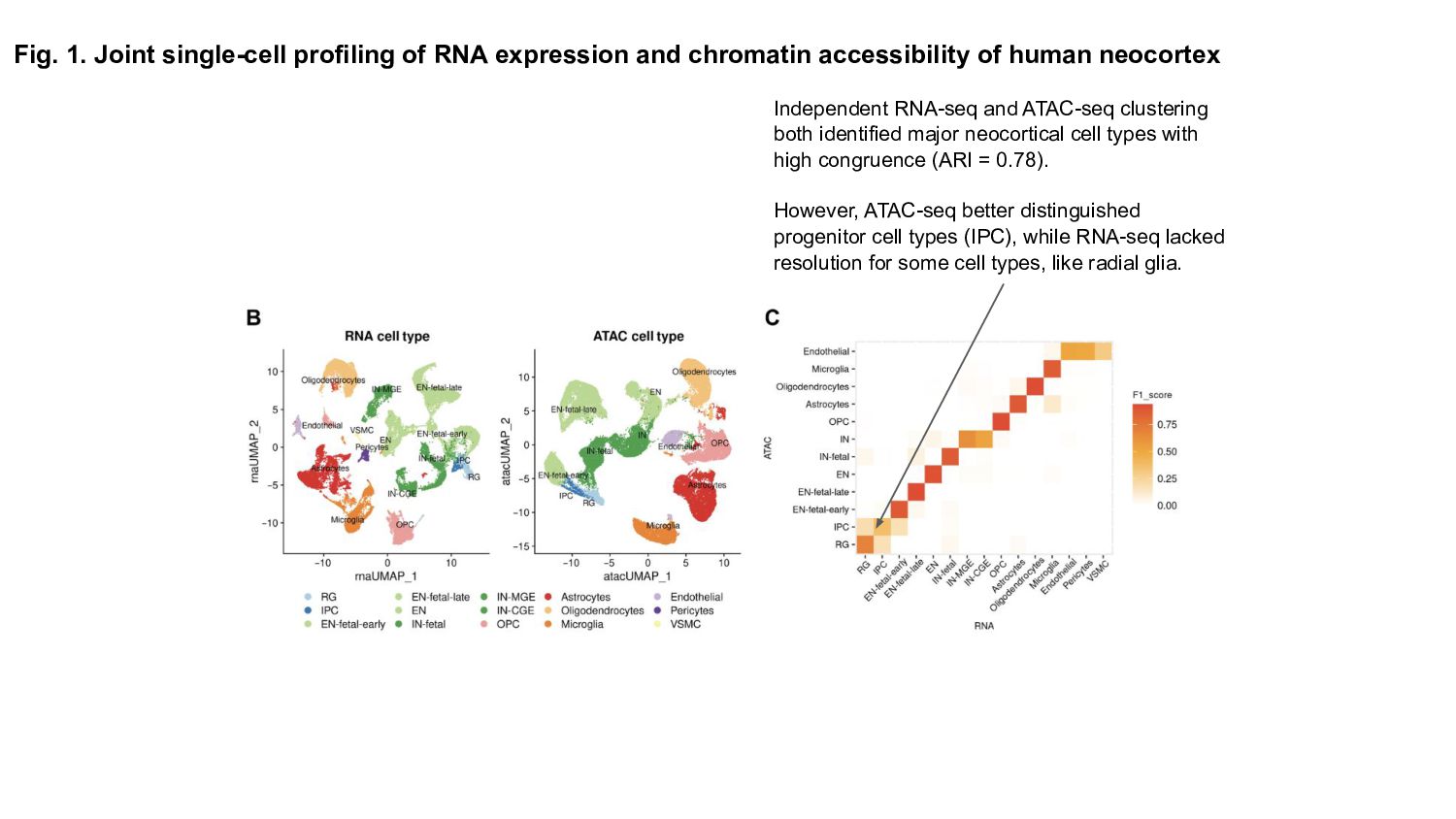
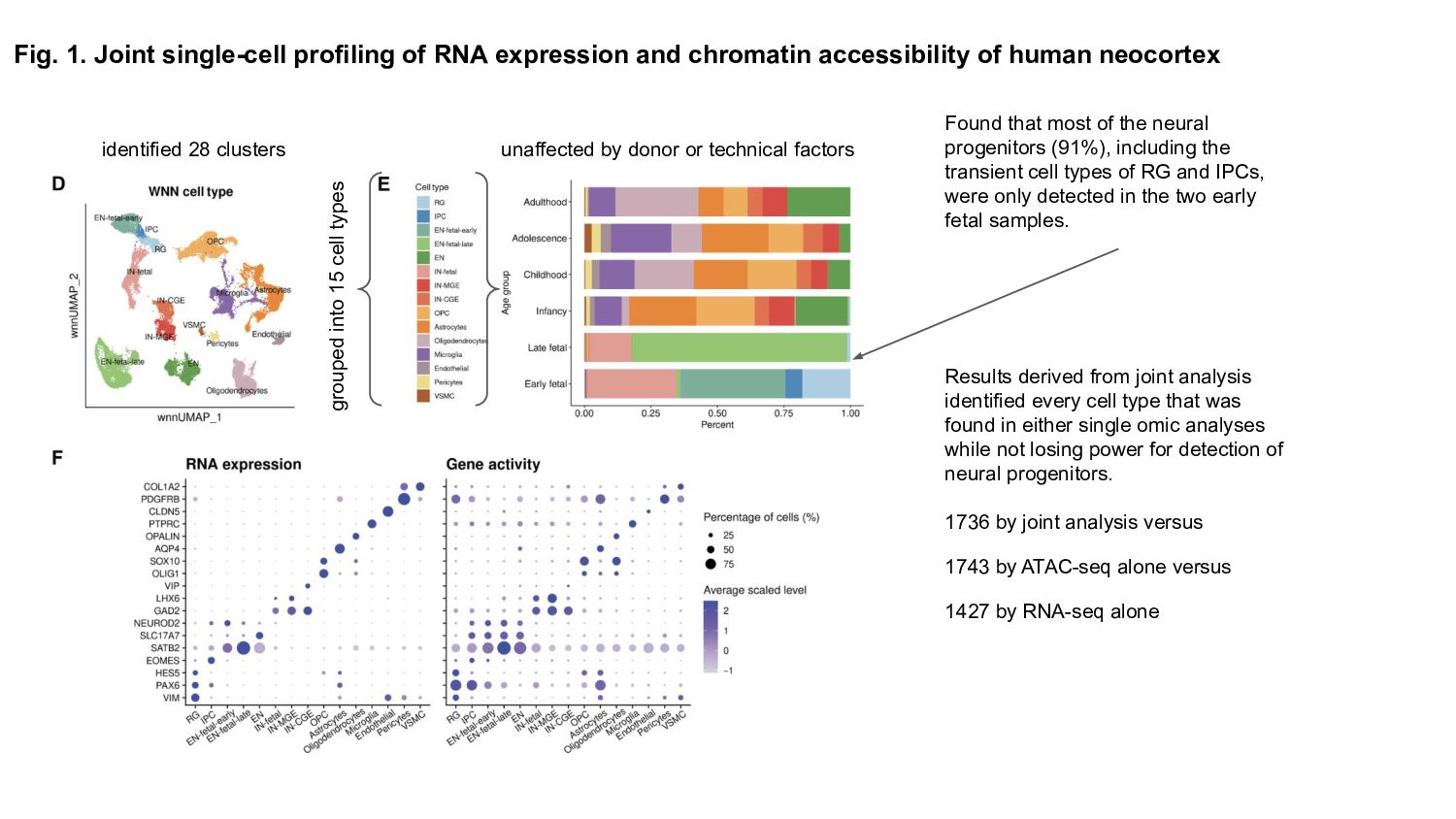
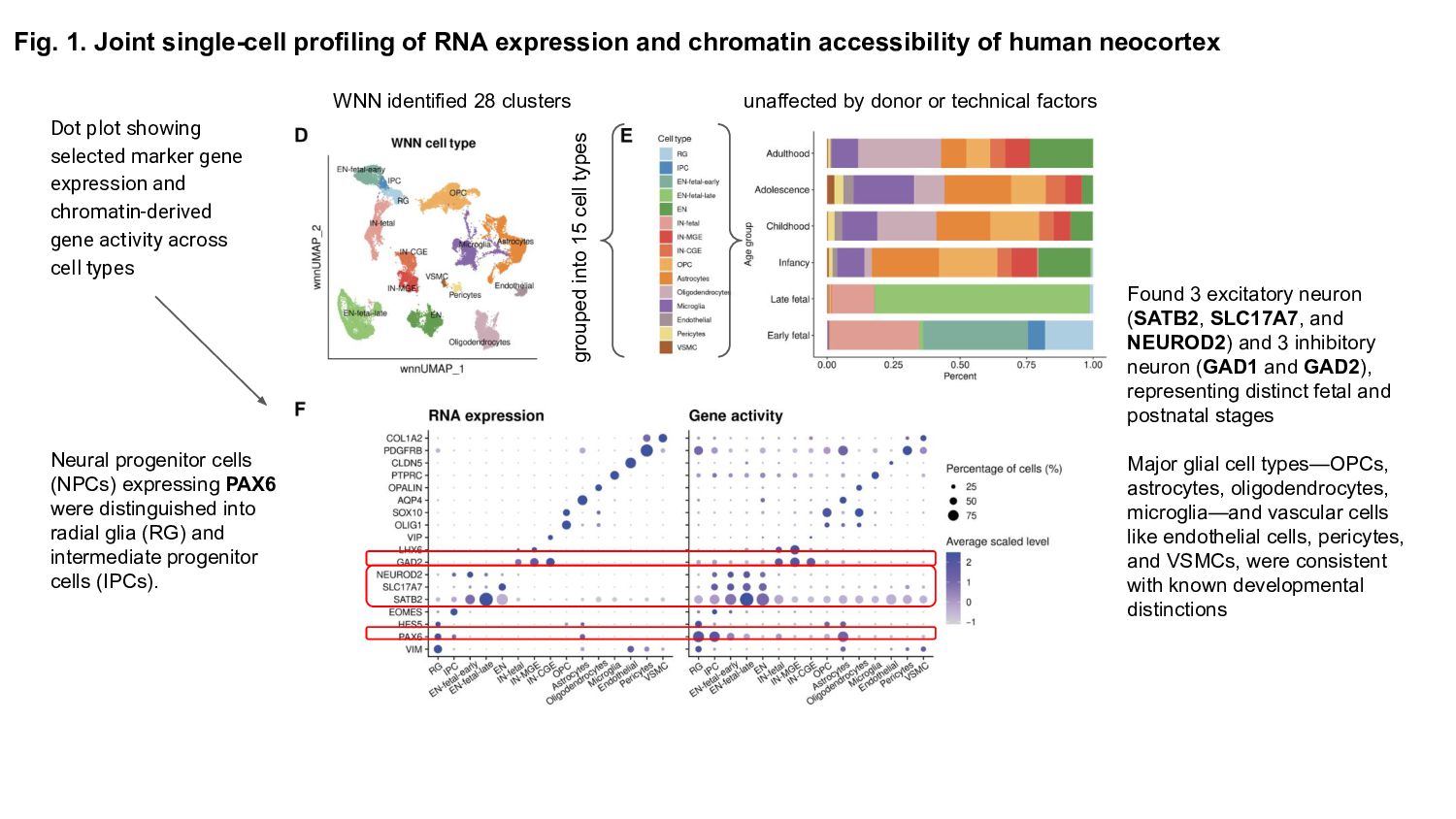
Prior studies have explored gene expression and epigenetic changes separately, this study combines single-cell gene expression and chromatin accessibility profiling across 45,549 cells at multiple developmental stages.
Background: Briefly introduce the significance of the human brain’s cellular complexity and the role of gene expression in development.
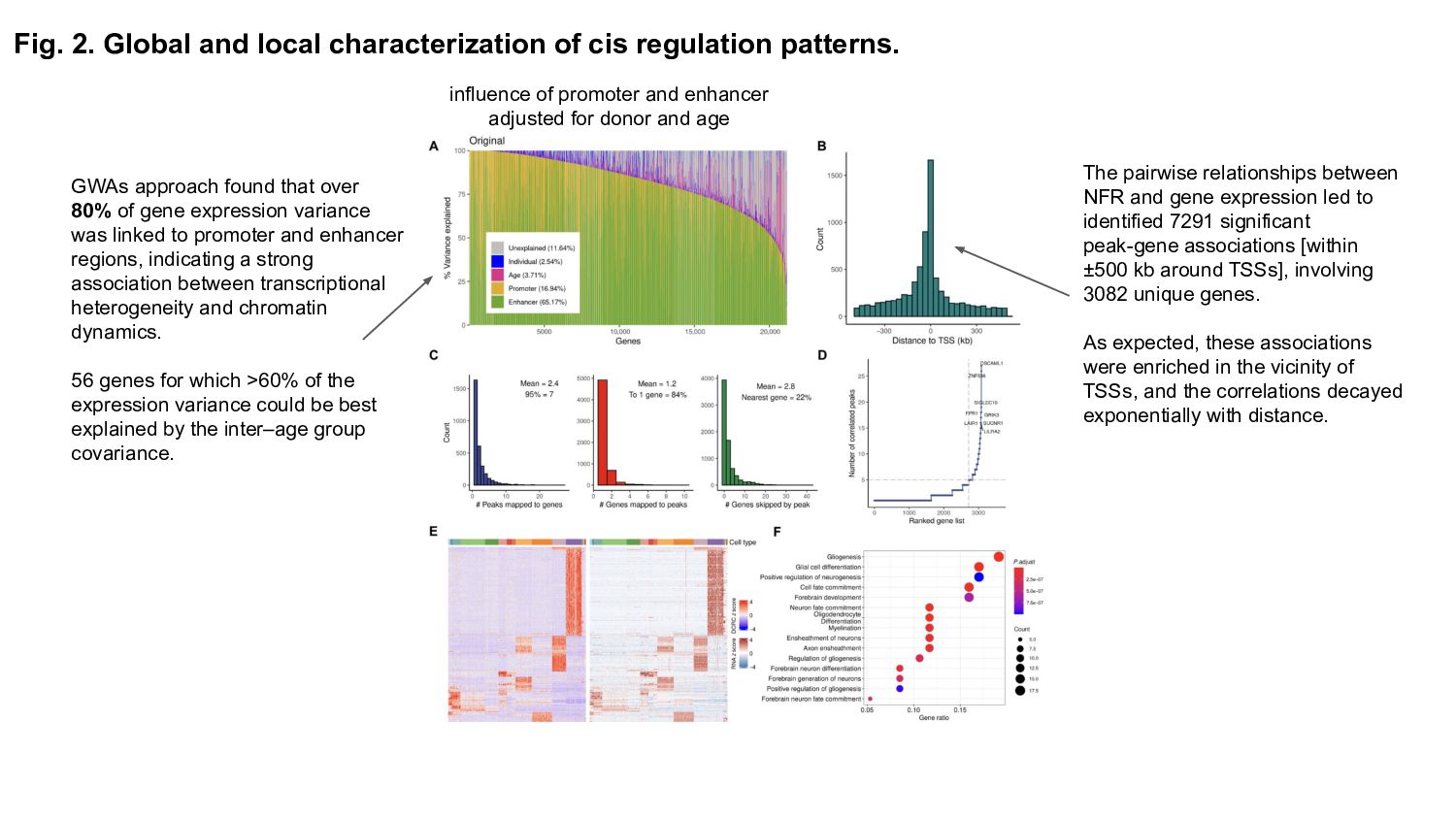
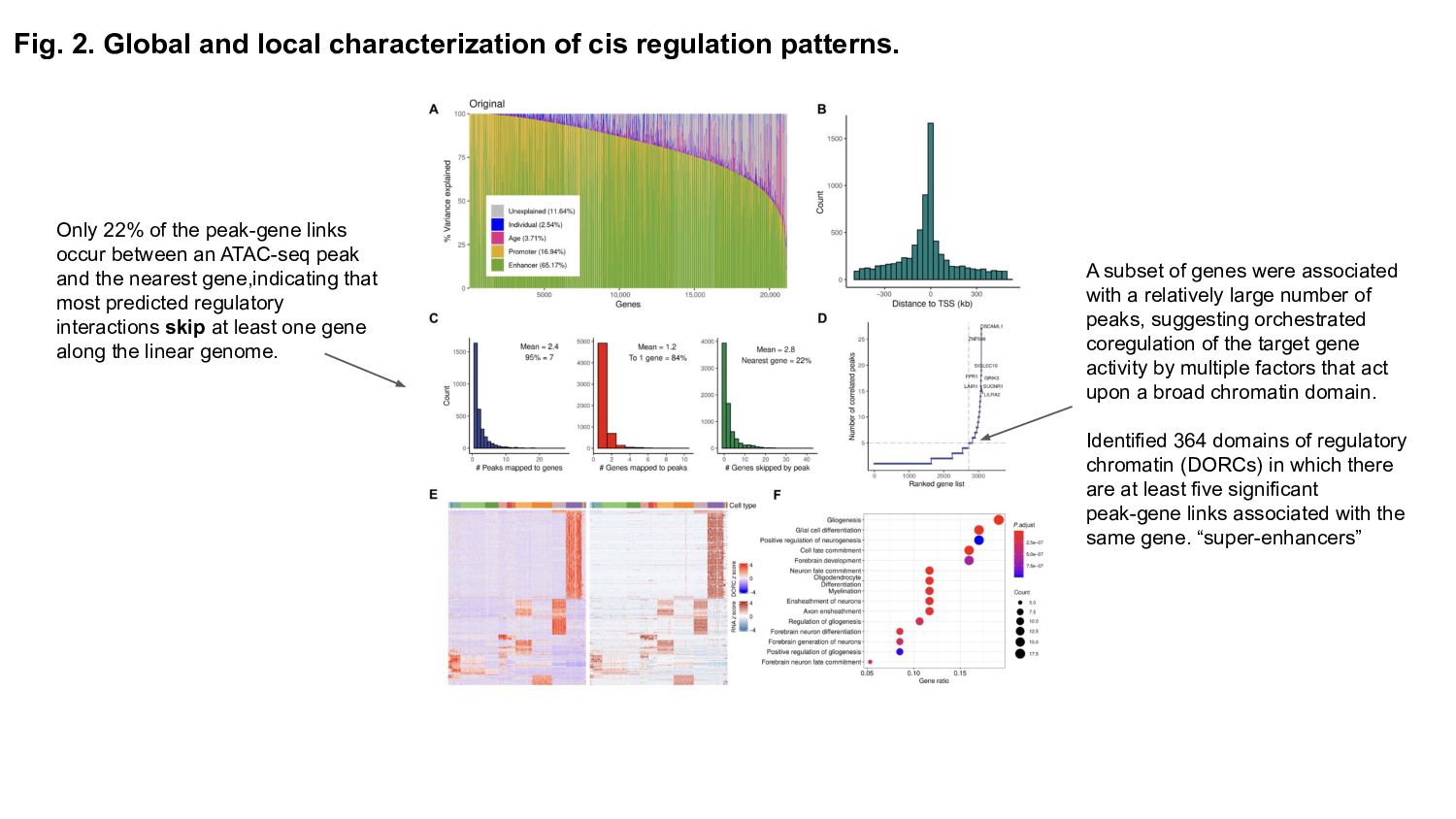
Role of CREs: Explain what cis-regulatory elements are and why their spatiotemporal activity is crucial for gene expression regulation during brain development.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}