4. Spots excluded from analysis for having a low library size as identified by scran for all 30 samples across the anterior-posterior axis of the DLPFC
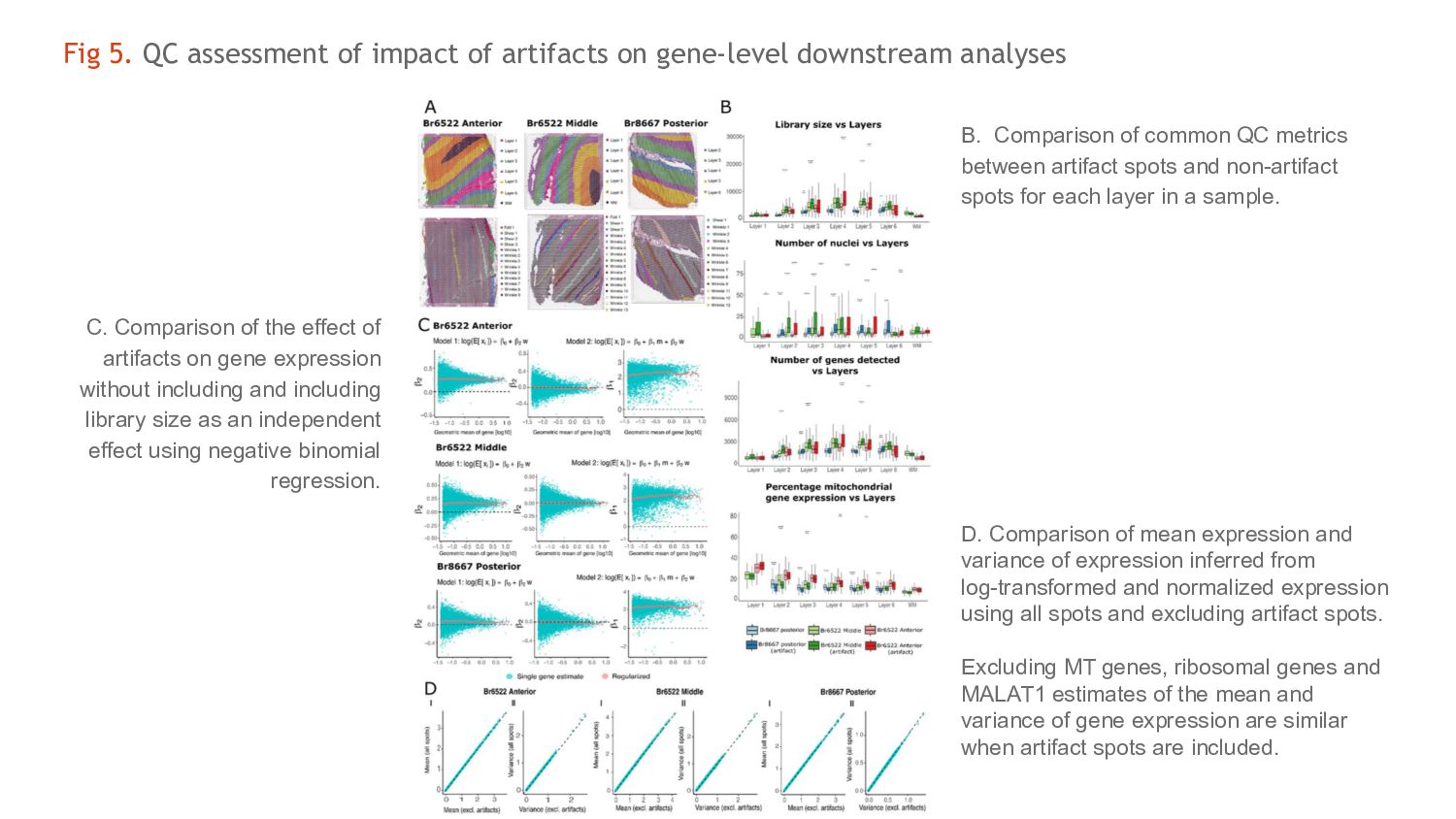
downstream analyses B. Comparison of common QC metrics between artifact spots and non-artifact spots for each layer in a sample. C. Comparison of the effect of artifacts on gene expression without including and including library size as an independent effect using negative binomial regression. D. Comparison of mean expression and variance of expression inferred from log-transformed and normalized expression using all spots and excluding artifact spots. Excluding MT genes, ribosomal genes and MALAT1 estimates of the mean and variance of gene expression are similar when artifact spots are included.
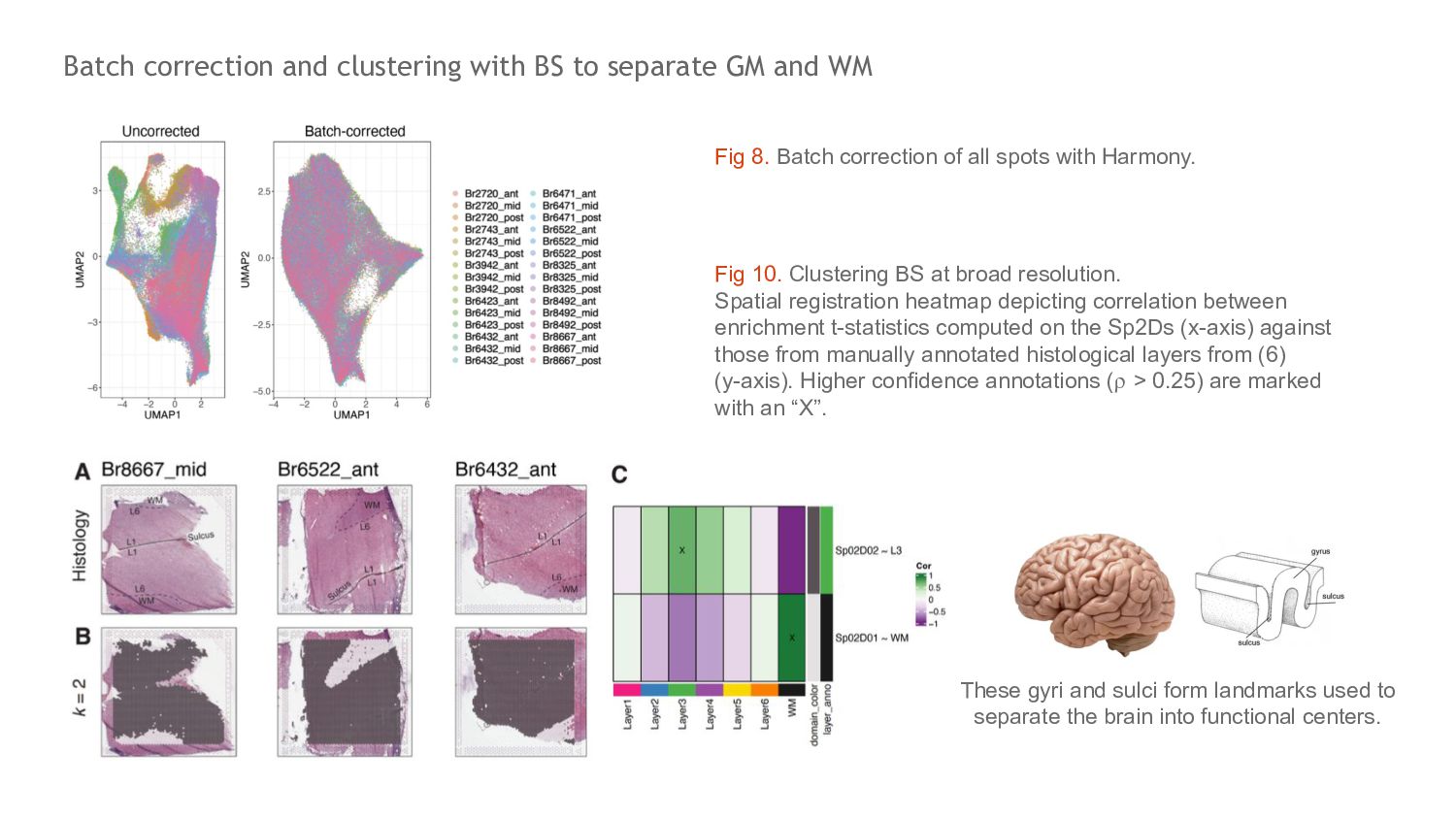
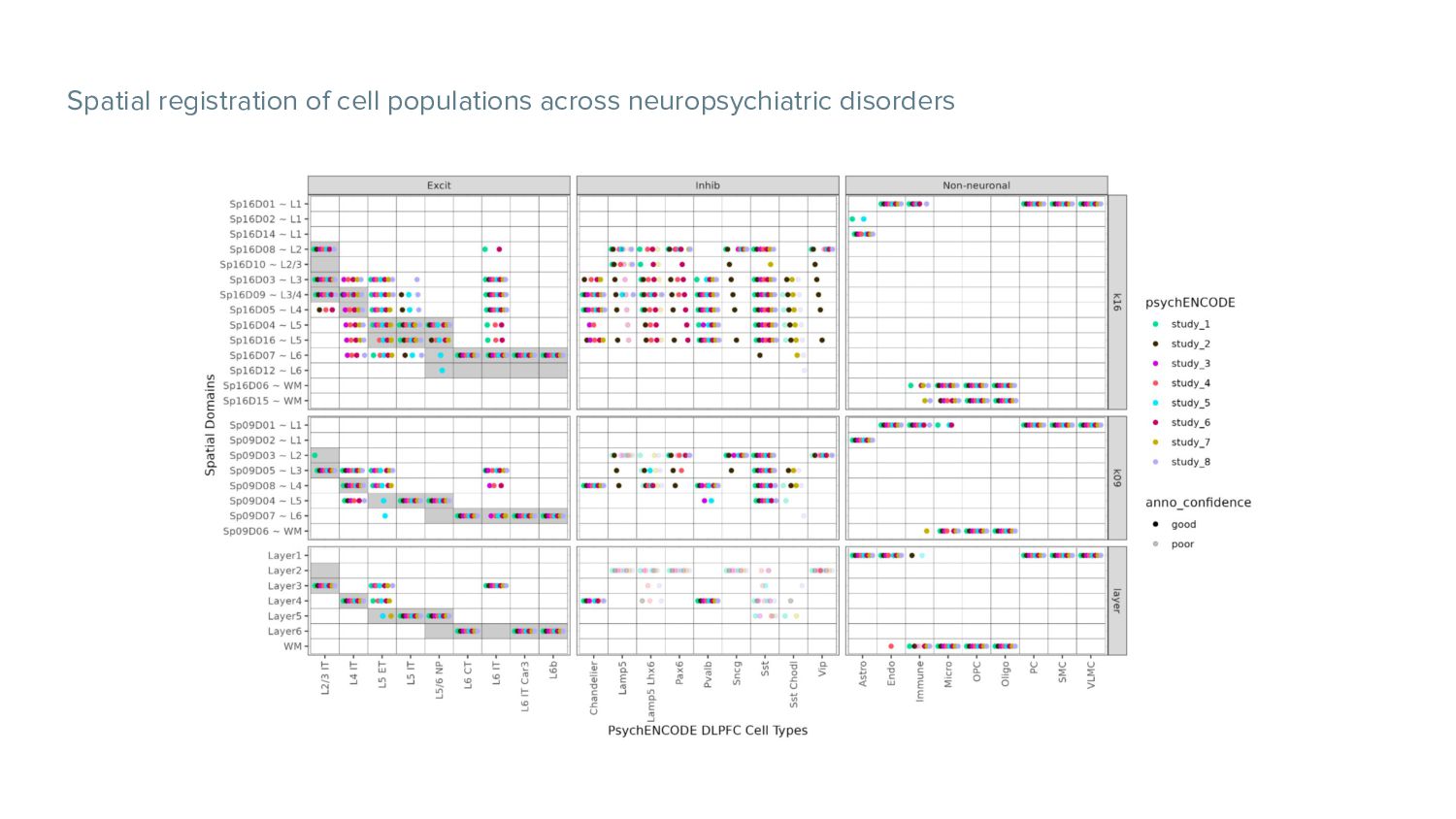
WM Fig 8. Batch correction of all spots with Harmony. Fig 10. Clustering BS at broad resolution. Spatial registration heatmap depicting correlation between enrichment t-statistics computed on the Sp2Ds (x-axis) against those from manually annotated histological layers from (6) (y-axis). Higher confidence annotations (⍴ > 0.25) are marked with an “X”. These gyri and sulci form landmarks used to separate the brain into functional centers.
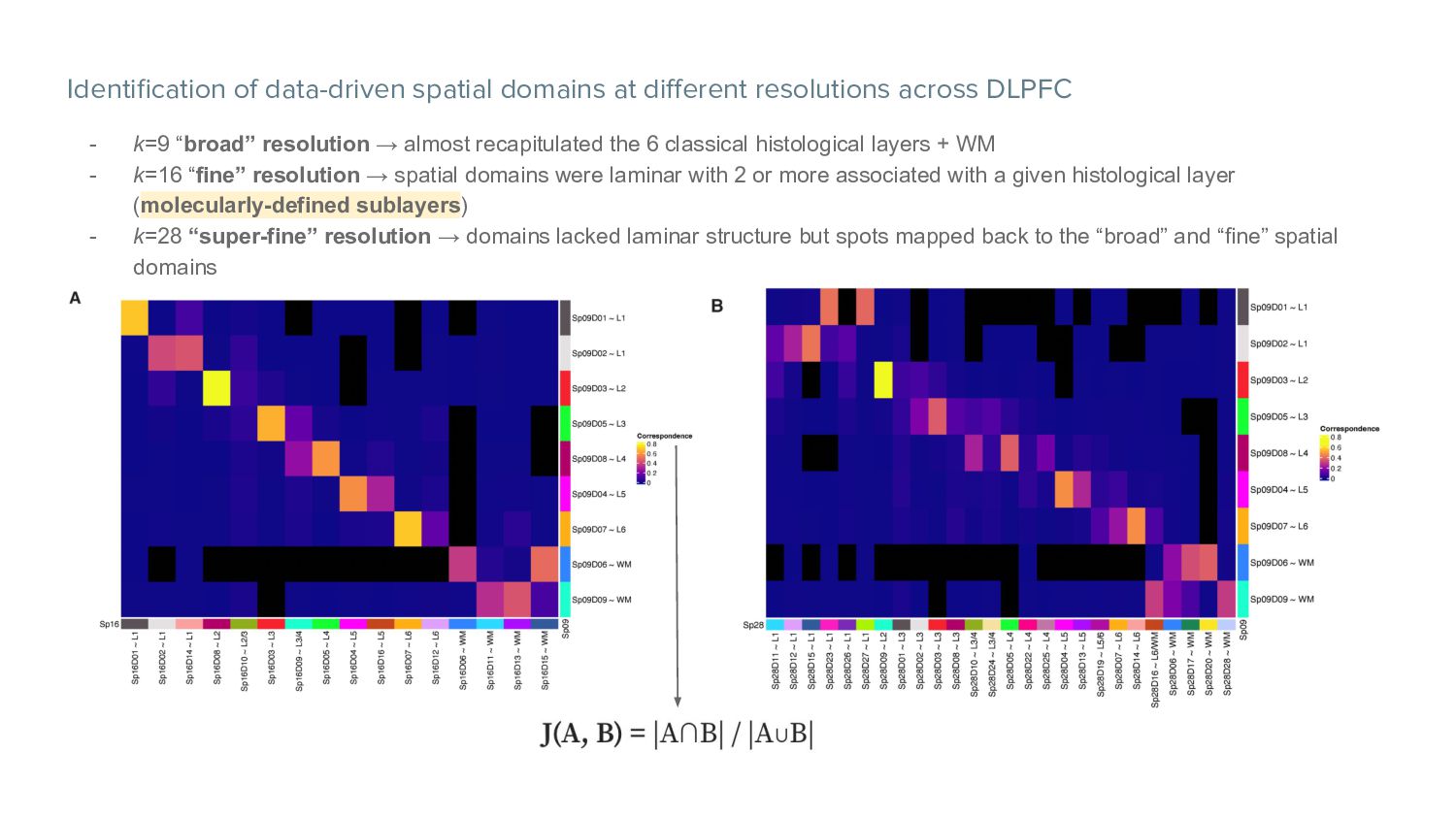
- k=9 “broad” resolution → almost recapitulated the 6 classical histological layers + WM - k=16 “fine” resolution → spatial domains were laminar with 2 or more associated with a given histological layer (molecularly-defined sublayers) - k=28 “super-fine” resolution → domains lacked laminar structure but spots mapped back to the “broad” and “fine” spatial domains
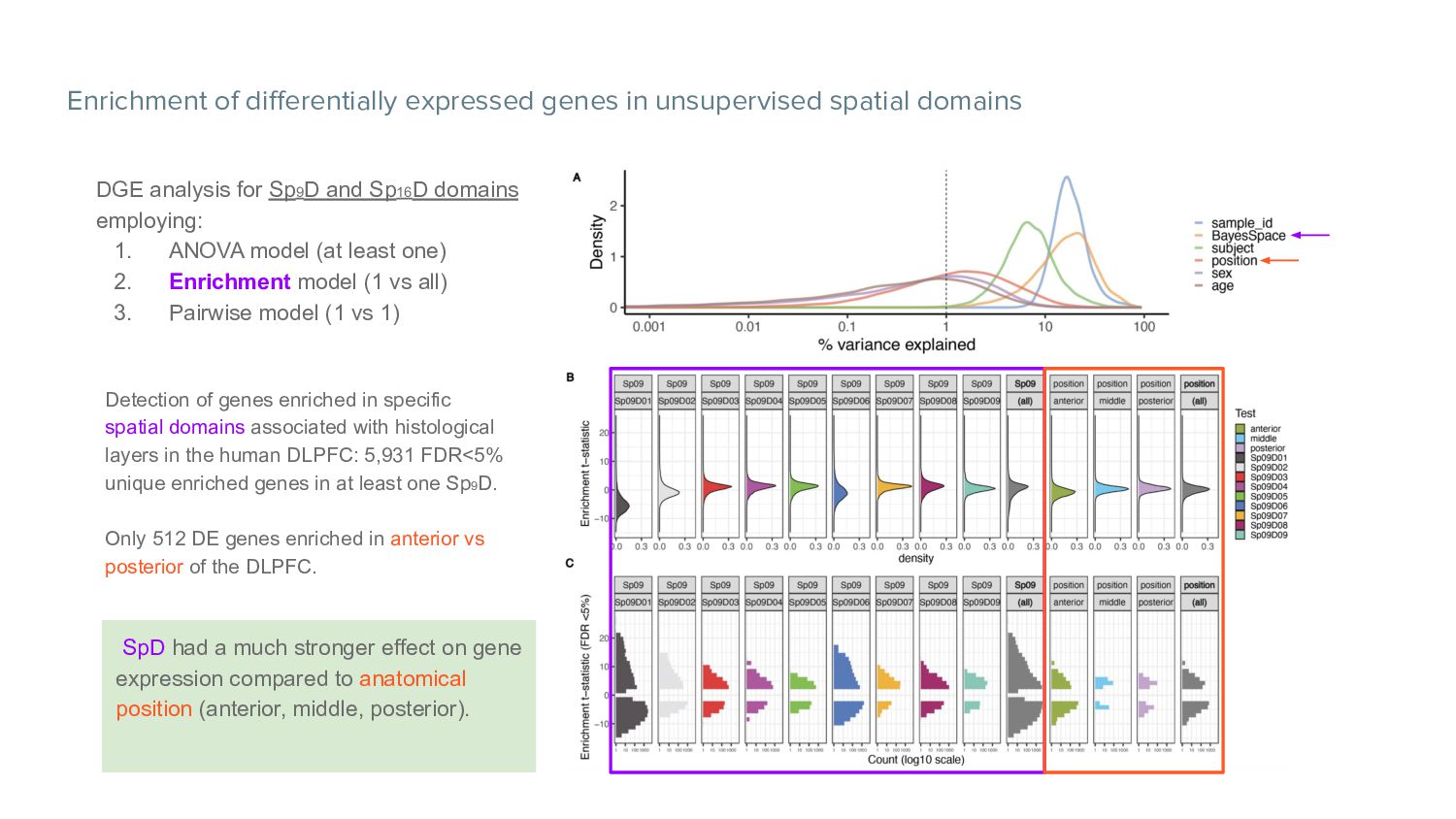
analysis for Sp9D and Sp16D domains employing: 1. ANOVA model (at least one) 2. Enrichment model (1 vs all) 3. Pairwise model (1 vs 1) Detection of genes enriched in specific spatial domains associated with histological layers in the human DLPFC: 5,931 FDR<5% unique enriched genes in at least one Sp9D. Only 512 DE genes enriched in anterior vs posterior of the DLPFC. SpD had a much stronger effect on gene expression compared to anatomical position (anterior, middle, posterior).
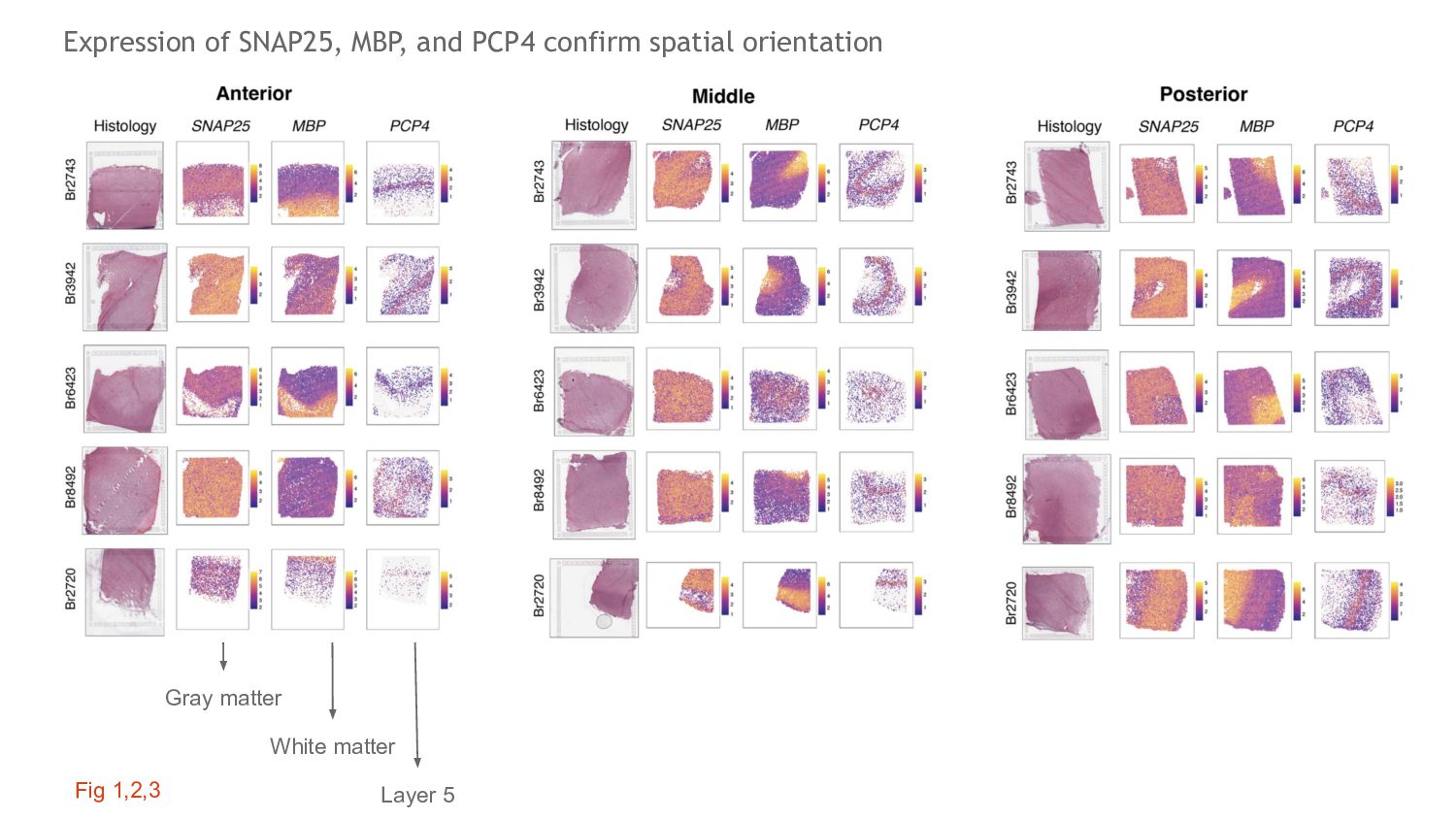
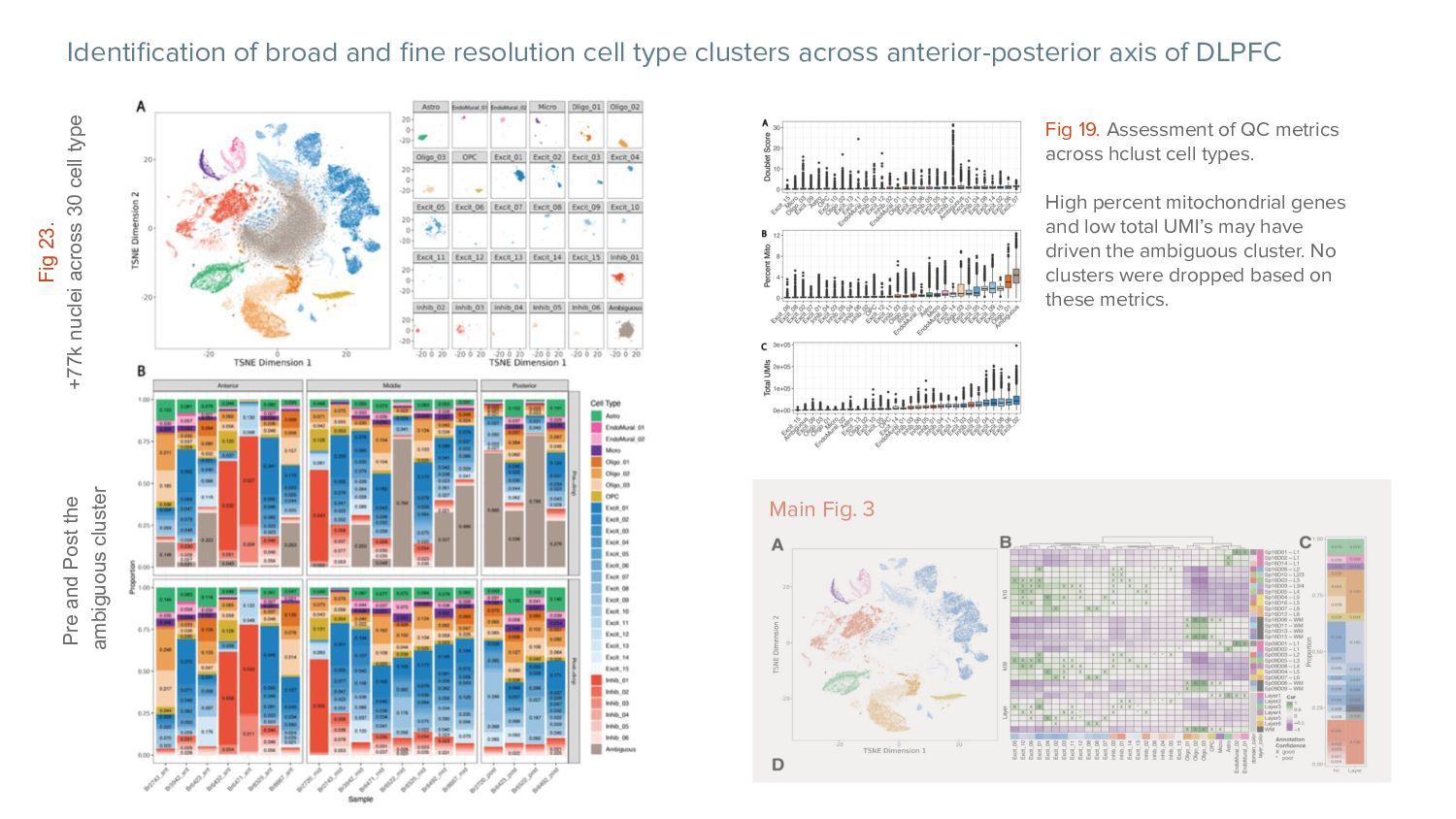
anterior-posterior axis of DLPFC Main Fig. 3 Fig 23. +77k nuclei across 30 cell type Fig 19. Assessment of QC metrics across hclust cell types. High percent mitochondrial genes and low total UMI’s may have driven the ambiguous cluster. No clusters were dropped based on these metrics. Pre and Post the ambiguous cluster
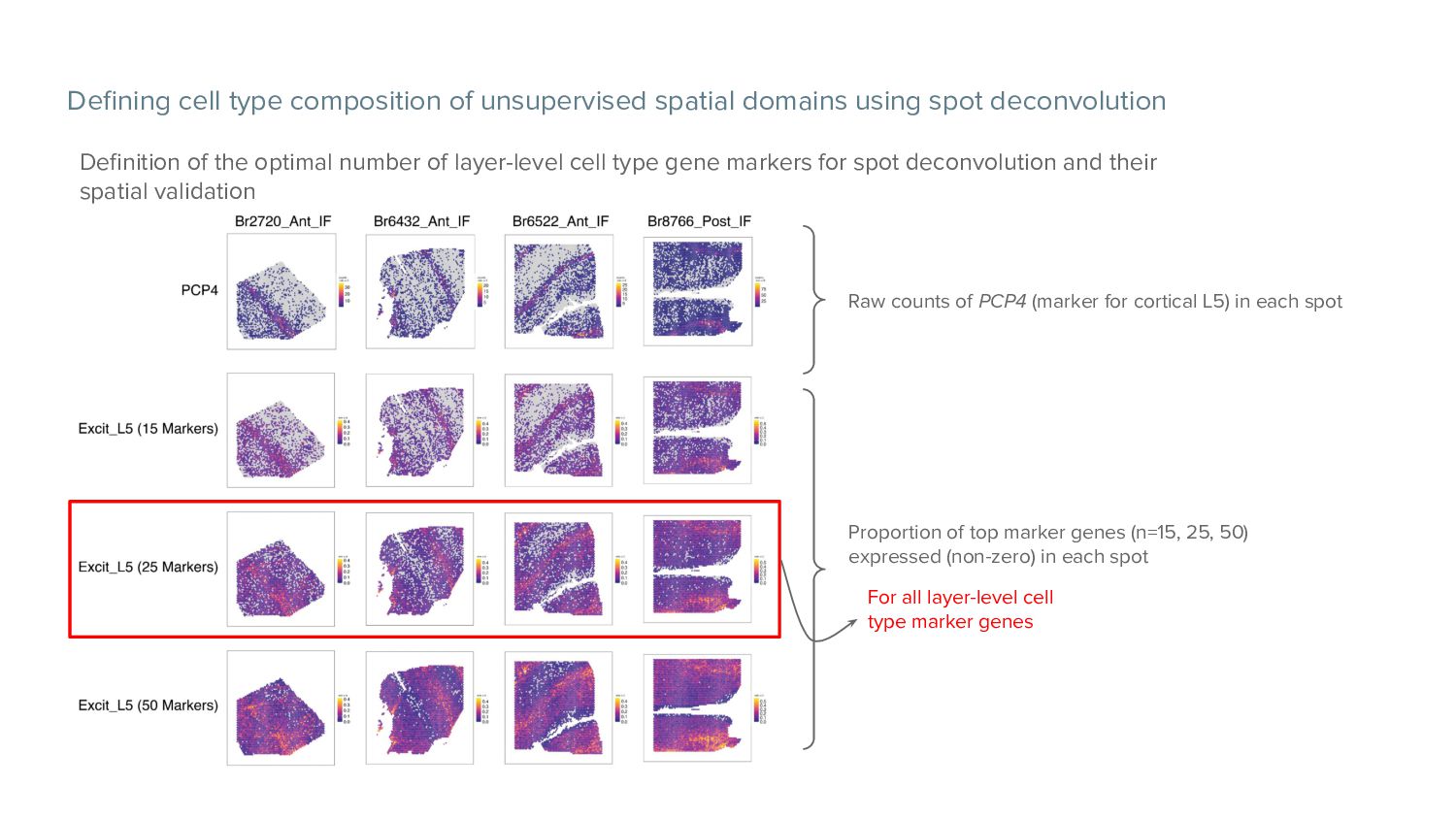
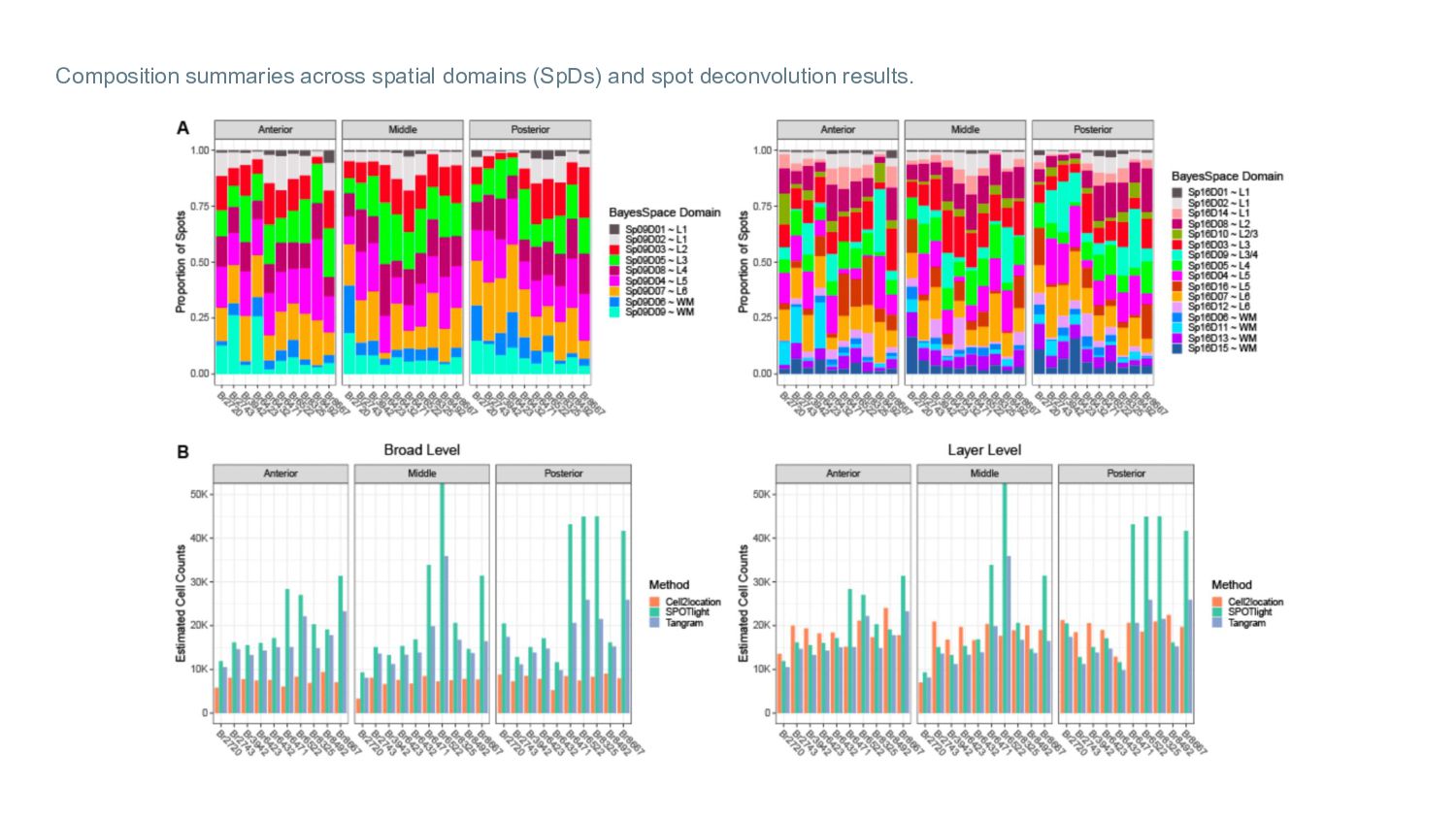
deconvolution Definition of the optimal number of layer-level cell type gene markers for spot deconvolution and their spatial validation Proportion of top marker genes (n=15, 25, 50) expressed (non-zero) in each spot Raw counts of PCP4 (marker for cortical L5) in each spot For all layer-level cell type marker genes
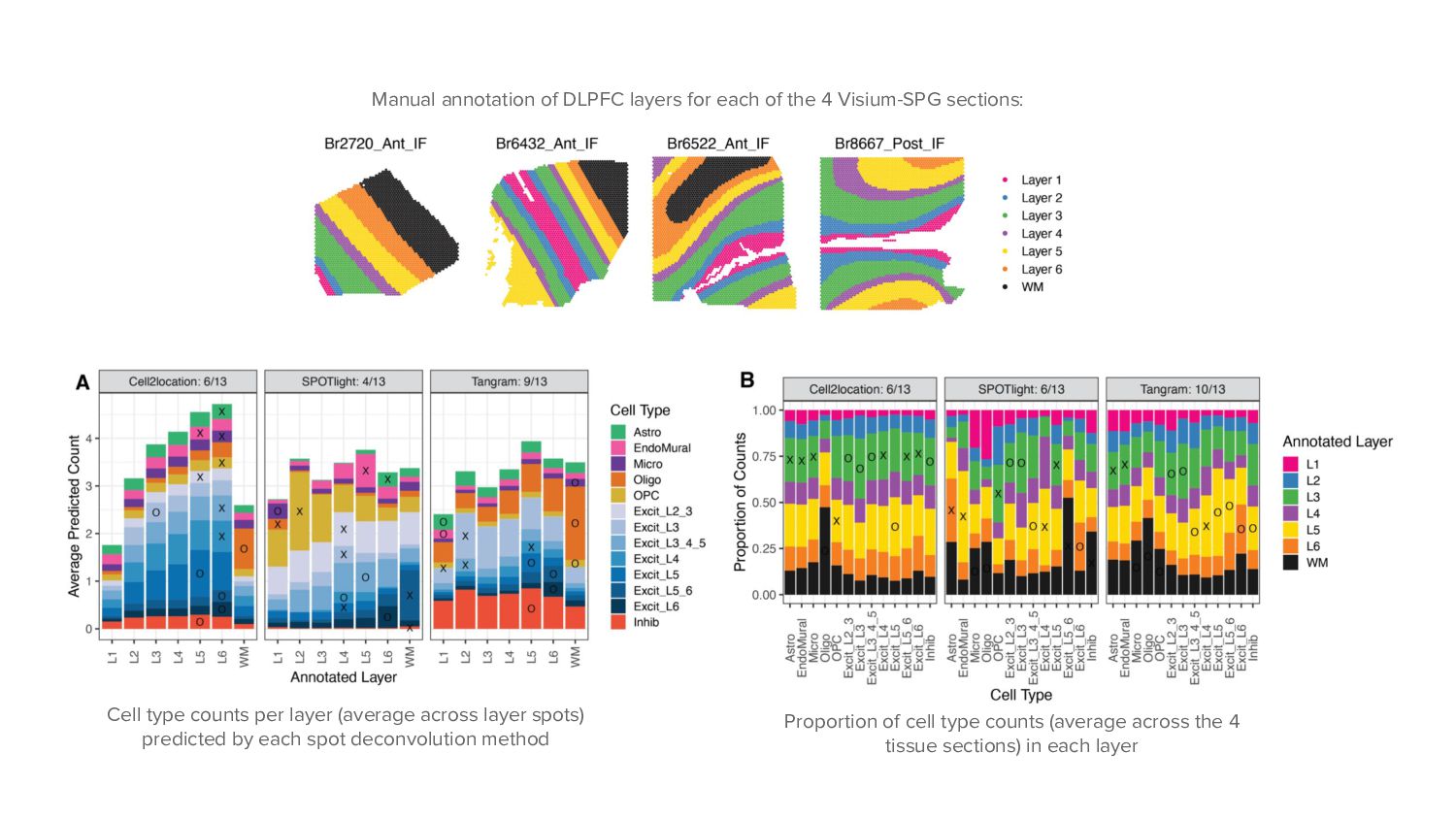
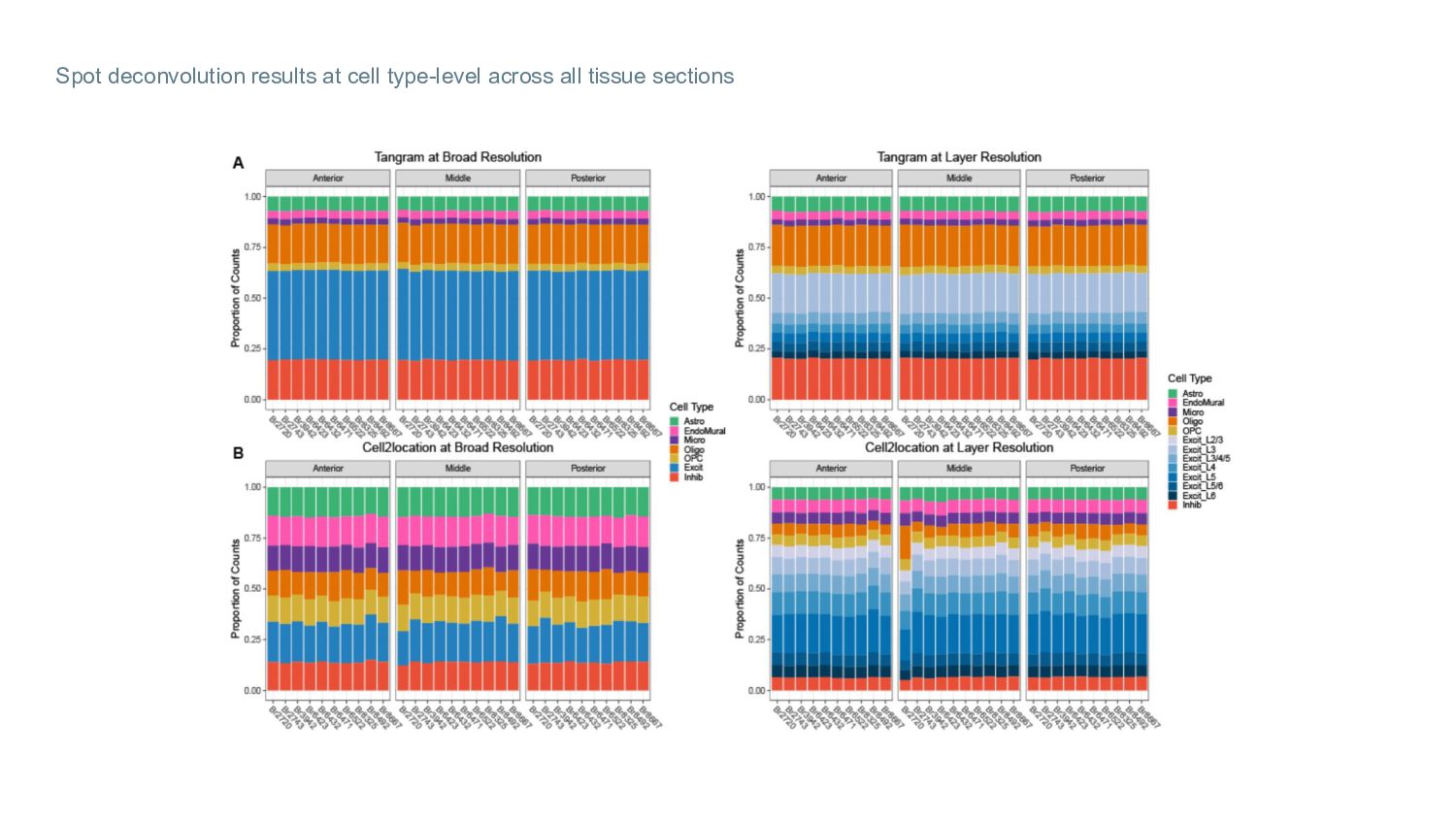
Visium-SPG sections: Cell type counts per layer (average across layer spots) predicted by each spot deconvolution method Proportion of cell type counts (average across the 4 tissue sections) in each layer
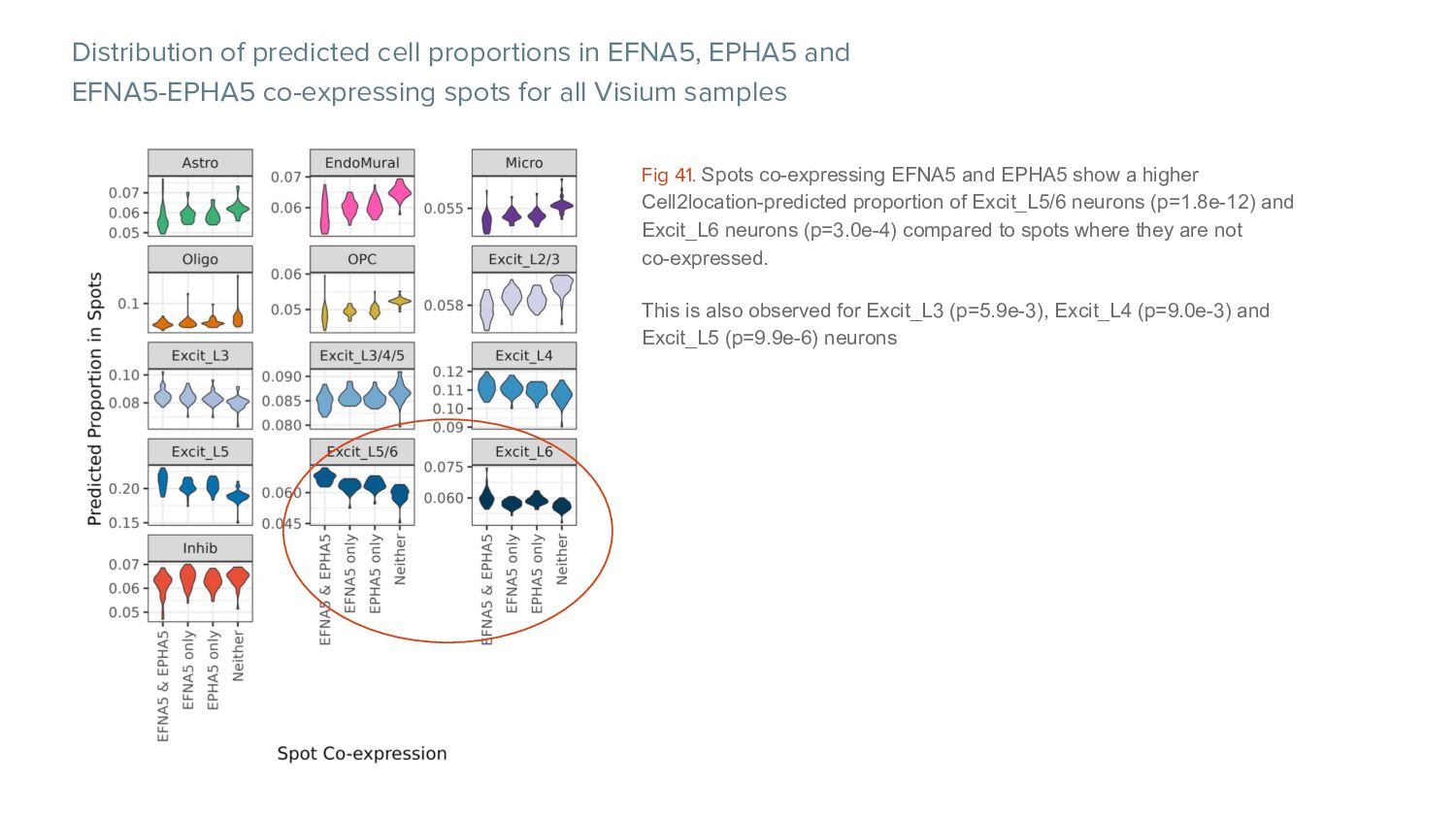
co-expressing spots for all Visium samples Fig 41. Spots co-expressing EFNA5 and EPHA5 show a higher Cell2location-predicted proportion of Excit_L5/6 neurons (p=1.8e-12) and Excit_L6 neurons (p=3.0e-4) compared to spots where they are not co-expressed. This is also observed for Excit_L3 (p=5.9e-3), Excit_L4 (p=9.0e-3) and Excit_L5 (p=9.9e-6) neurons
the human prefrontal cortex revealing novel spatial domains and cell-cell interactions relevant for psychiatric diseases. • They used Visium spatial technology integrated with snRNA-seq data to identified distinct cell types and to discover cell-cell interactions across spatial domains. • They used PsychENCODE and publicly data to map the enrichment of cell types and genes associated with neuropsychiatric disorders to the spatial domains • They integrated spatial and snRNA-seq datasets are available at research.libd.org/spatialDLPFC/ shiny app to be explored for the scientific community.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}