Sequence Matrix[1] is a freely-available, cross-platform application that lets you concatenate gene datasets easily. A spreadsheet-like interface displays what you've assembled so far; TNT, Nexus, FASTA and Mega files can be dragged into the application to add them. Your entire dataset can be exported as TNT or Nexus files.
This presentation describes Sequence Matrix, why you might want to use it, and how it can help you prepare large, multigene/multitaxon datasets for easy analysis.
[1] http://code.google.com/p/sequencematrix/
{kind=link}
{kind=link}
{kind=link}
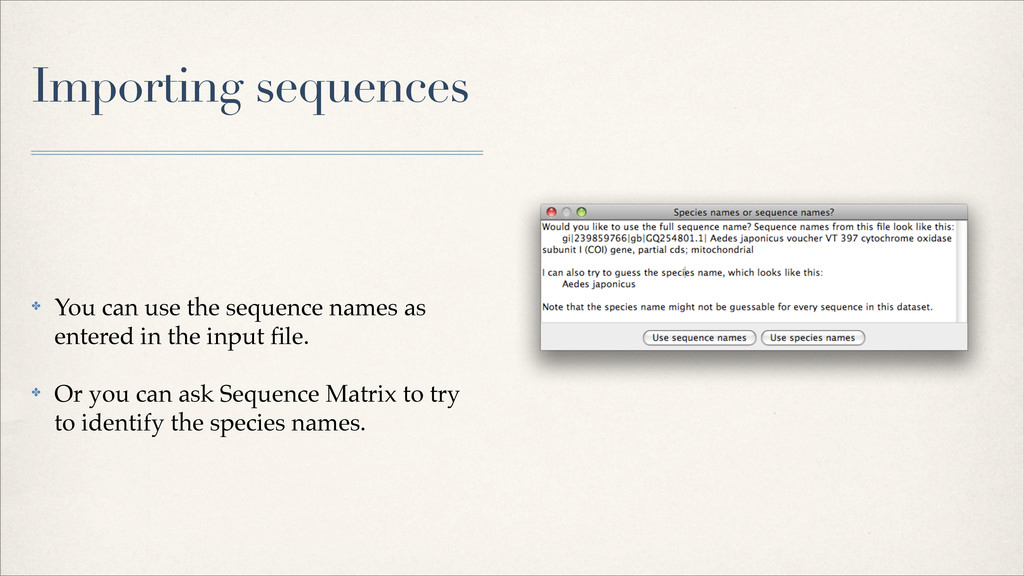{kind=link}
{kind=link}
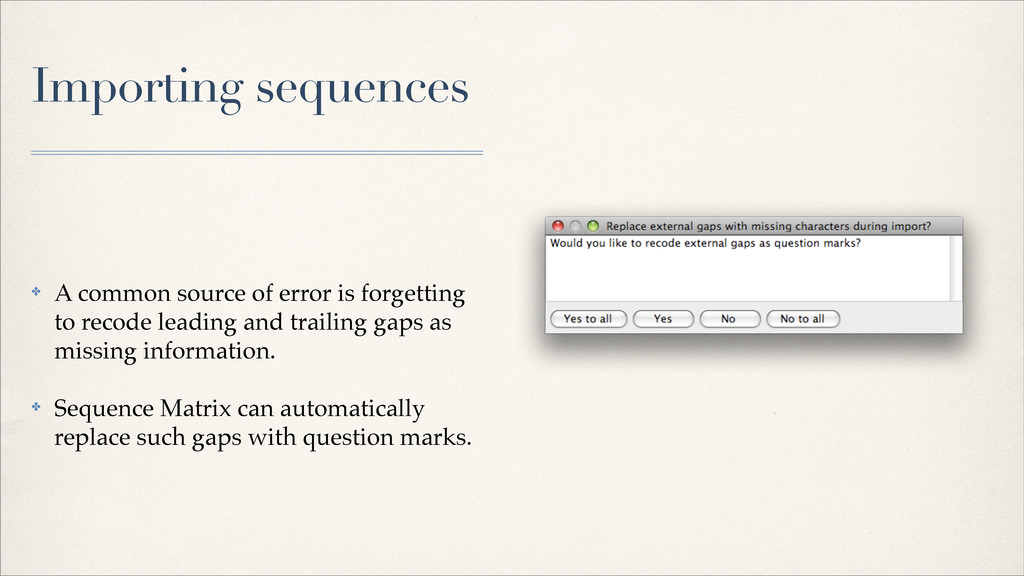{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
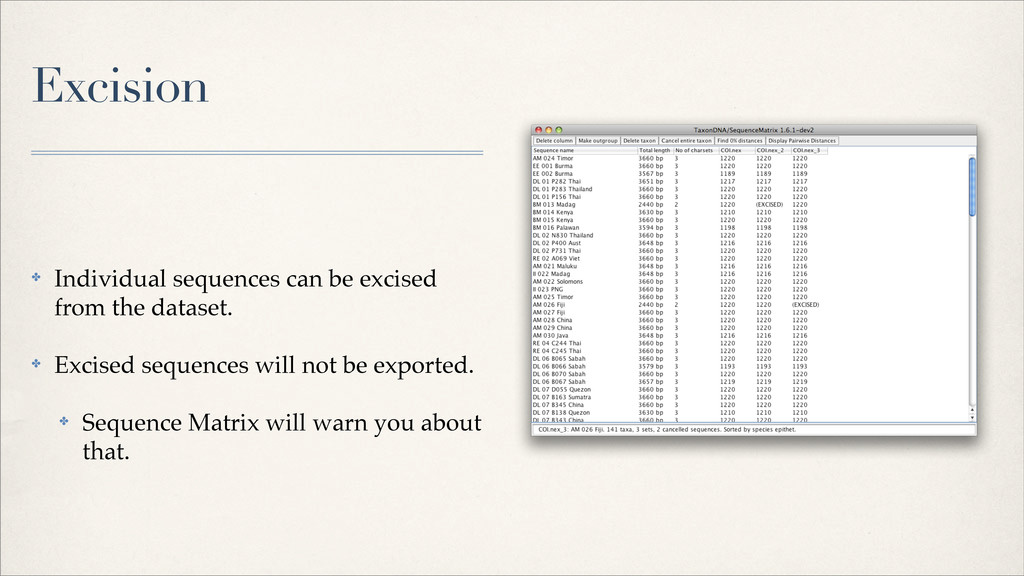{kind=link}
{kind=link}
{kind=link}
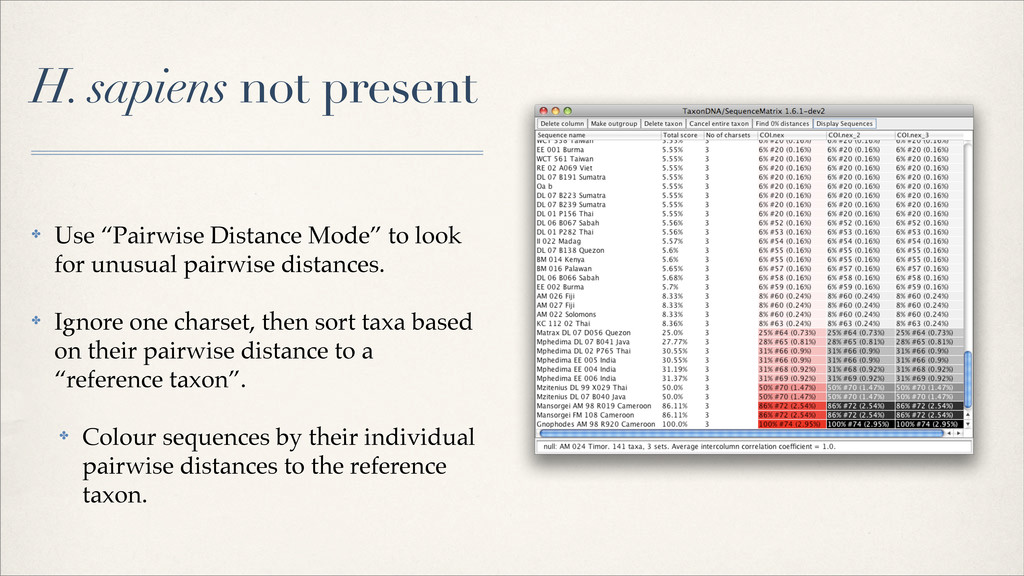{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}