DNA fragments. 2. Sequence the DNA fragments. 3. Connect the abundance of fragments to the rate of the biological process. Typically named as: SOMETHING-Seq Examples: ChIP-Seq, RNA-Seq, RAD-Seq All work by co-opting an existing biological mechanism to produce DNA fragments.
biological function. Thus the abundance of fragments ought to correlate with the activity level of the function. There are ongoing debates on the range of validity of each approach. We "force" the biological function to produce DNA fragments that we can sequence.
what a function assay does. Example: "ChIP-Seq measures the rate of protein binding to DNA." Reality is more complicated: "ChIP-Seq measures how frequent DNA fragments are. Bound DNA locations produce more fragments than unbound locations"
to understand in what way does the data deviate from random DNA fragmentation. 1. Understand what gets sequenced. 2. Understand what can and cannot be quanti ed. Both can be suprisingly challenging get right - a typical mistake is to assume more than what a method does in reality.
types: rRNA , mRNA , tRNA , snoRNA , miRNA , etc. mRNA needs to be puri ed from total RNA. RNA needs to be reverse transcribed into DNA. Transcripts express at very different levels. Isoforms may be very similar? What detection level? Is the strand information retained? Each step introduces its own biases and challenges.
similar (perhaps with little twists). Align Quantify Compare Interpreting the results is different. Now the speci cs of the origin of the DNA matter A LOT. Understanding a *Seq method means understaning the origin of the DNA.
Coverage is the numer of reads that "cover" something. Base coverage: ---- ------ ------- ----- ----- ---- 0112211211222232211110 Coverage: how many sequencing reads cover a base.
A ==| What is the coverage of transcript A? Is it 3 , 4 or 5 ? Depends on what "counting strategy" we use 3 if all reads must be fully inside. 4 if at least 50% of a read must be inside. 5 if any overlap counts as a coverage.
compare across different conditions: 1. Compute the coverage over a thing in condition 1 --> it produces a number: 100 2. Compute the coverage over the same thing in condition 2 --> it produces another number: 200 Do we have suf cient evidence that the two numbers represent a change?
the same condition: 1. Compute the coverage over a thing in condition 1 --> it produces a number: 100 2. Compute the coverage over another thing in condition 1 --> it produces another number: 200 Do we have suf cient evidence that the two numbers represent a change?
useful skill applicable to many domains. There are RNA-Seq speci c approaches but it is important to understand how to do it in a generic way. Start with the megaton.sh that we used for Lecture 26. This is why is good to have megaton scripts lying around. http://data.biostarhandbook.com/variant/megaton.sh bash megaton.sh
or more ranges with the in a chrom:start-end format: samtools view -c SRR1972739.bam AF086833:1-1000 Prints 141 You can combine the query with SAM ags as well: samtools view -c -f 16 SRR1972739.bam AF086833:1-1000 54
on annotations (interval les) of any kind bedtools might just be the most useful tool in your toolkit. bedtools: flexible tools for genome arithmetic and DNA sequence usage: bedtools <subcommand> [options] The bedtools sub-commands include: [ Genome arithmetic ] intersect Find overlapping intervals in various ways. window Find overlapping intervals within a window aro closest Find the closest, potentially non-overlapping coverage Compute the coverage over defined intervals. map Apply a function to a column for each overlapp genomecov Compute the coverage over an entire genome. merge Combine overlapping/nearby intervals into a si
with intervals. 1. Create new intervals based on existing ones. 2. Extract or invert intervals. 3. Select or count overlapping intervals. 4. Extract sequences from a genome based on intervals. bedtools undertstands both BED , GFF VCF and BAM les and makes use of them correctly.
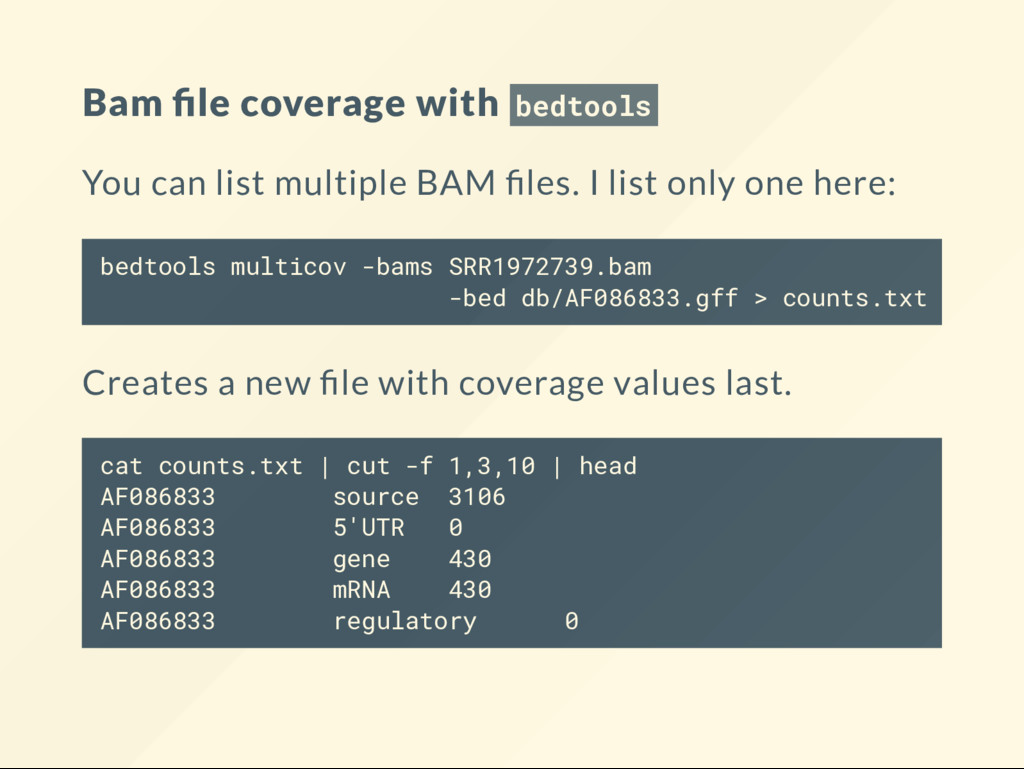
les. I list only one here: bedtools multicov -bams SRR1972739.bam -bed db/AF086833.gff > counts.txt Creates a new le with coverage values last. cat counts.txt | cut -f 1,3,10 | head AF086833 source 3106 AF086833 5'UTR 0 AF086833 gene 430 AF086833 mRNA 430 AF086833 regulatory 0
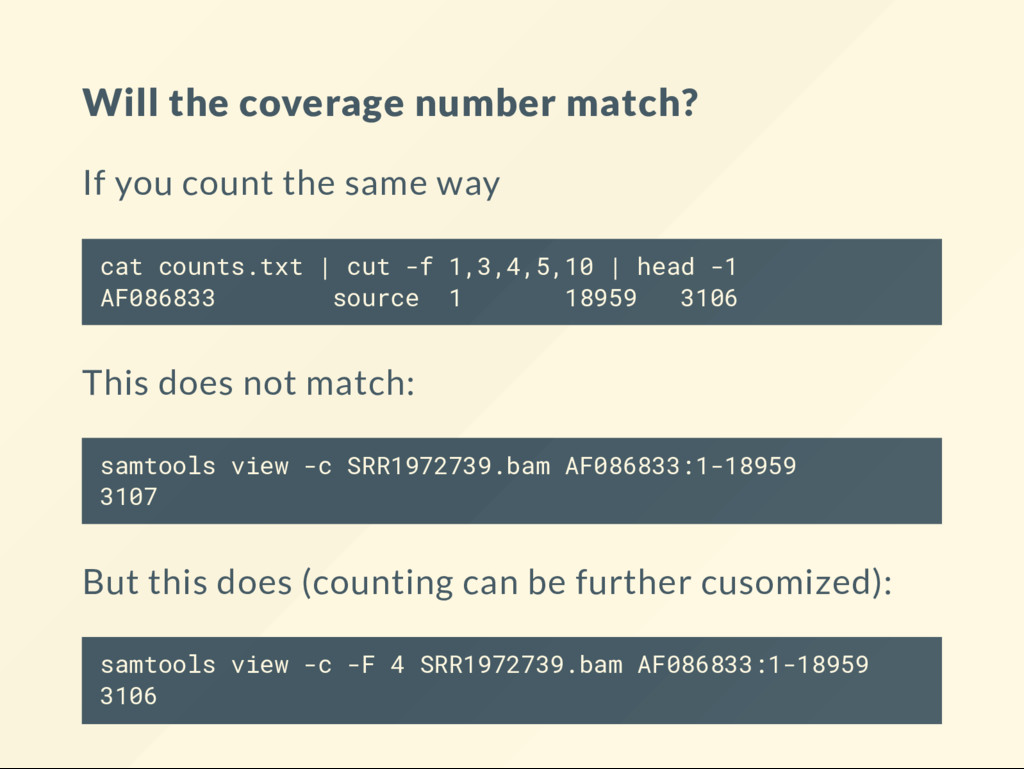
way cat counts.txt | cut -f 1,3,4,5,10 | head -1 AF086833 source 1 18959 3106 This does not match: samtools view -c SRR1972739.bam AF086833:1-18959 3107 But this does (counting can be further cusomized): samtools view -c -F 4 SRR1972739.bam AF086833:1-18959 3106
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}