Abstract
Atomistic simulation needs custom models, to approximate through hard O(N!) scaling of the underlying physical equations. Julia is a natural language to express such physical abstractions in. I will show how you can contribute to materials research with Julia and a fistful of 1960s PhysRevs.
Description
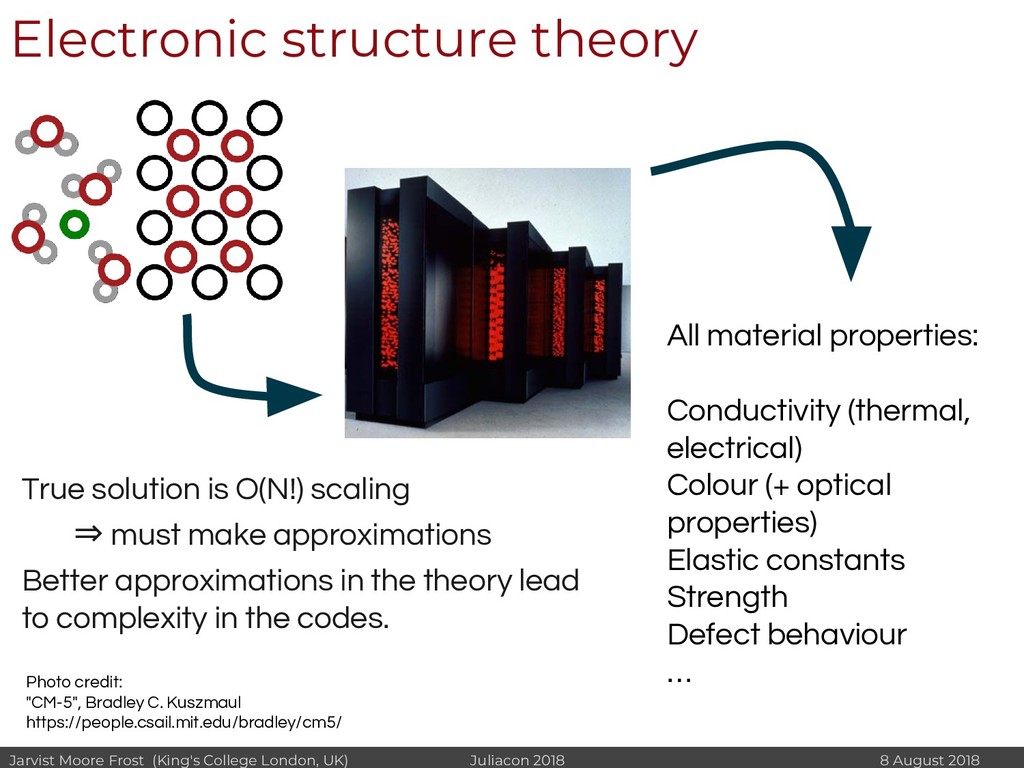
We can predict the properties of materials entirely within a computer. All material properties - strength, colour, opacity, conductivity etc. - come about as a result of the electronic structure adopted by the electrons in a material. This we can calculate by solving the Schrodinger equation. Unfortunately exact solutions are computationally intractable due to the O(N!) scaling with the number of electrons - the electron correlation problem. Progress in computational materials design has been made by solving approximate forms of this equation, numerically.
Venerable codes written in Fortran (and to a limited extent in C) consume the majority of cycles on public super computers. The often archaic code bases limits how easily they can be extended. The existence of standard, optimised, codes has enabled an enormous amount of research. At the same time, development of codes in relatively few walled gardens of physics has reduced the scope of techniques that are applied.
Modern materials design attempts to go beyond predicting properties of pure crystals. Generally this is because disordered materials can be made with less energy and time. Trying to predict the properties of these materials is a challenge, as the length and time scales so exceed that which we can get to even with approximate theories. As such, we need bespoke codes and models to be able to model these systems.
I will describe successful research projects in condensed matter theory which have made essential use of Julia, and will attempt to explain why Julia, with its mathematical expressiveness, physical abstraction, and high performance, is a natural language for future work in condensed matter theory.
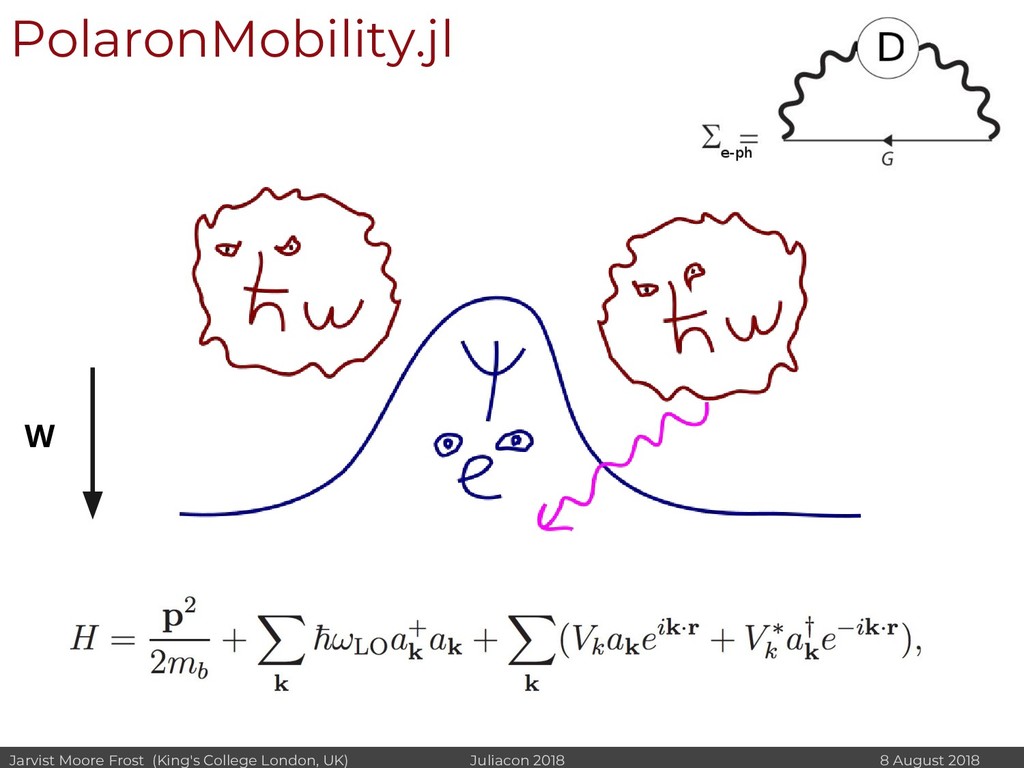
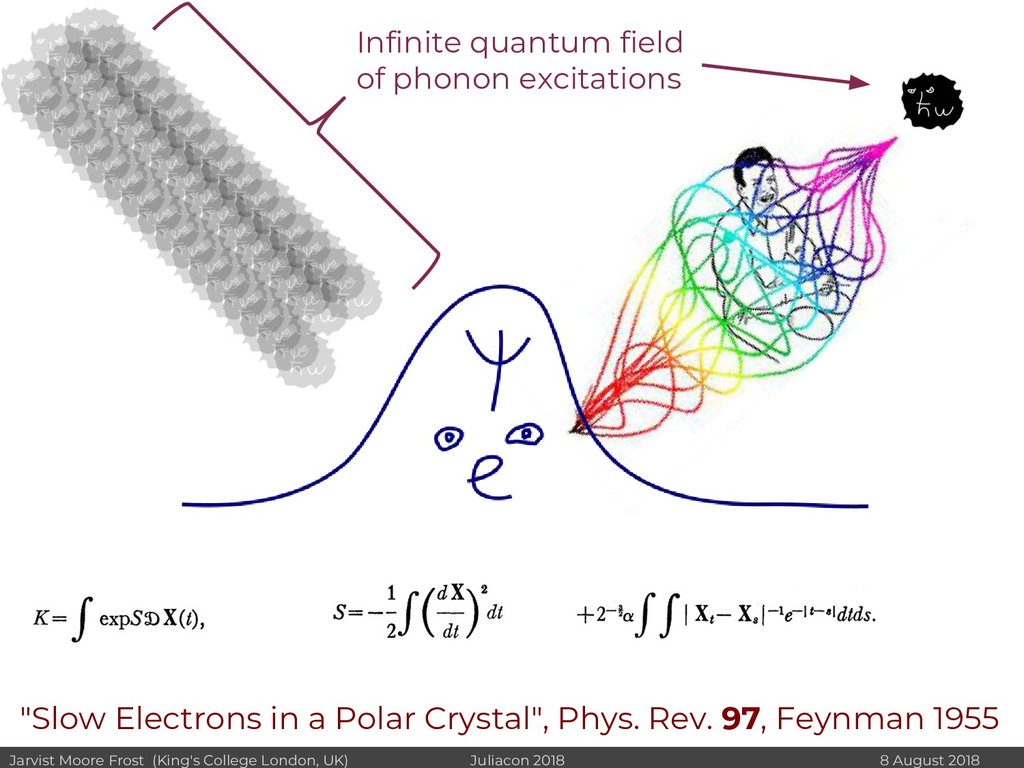
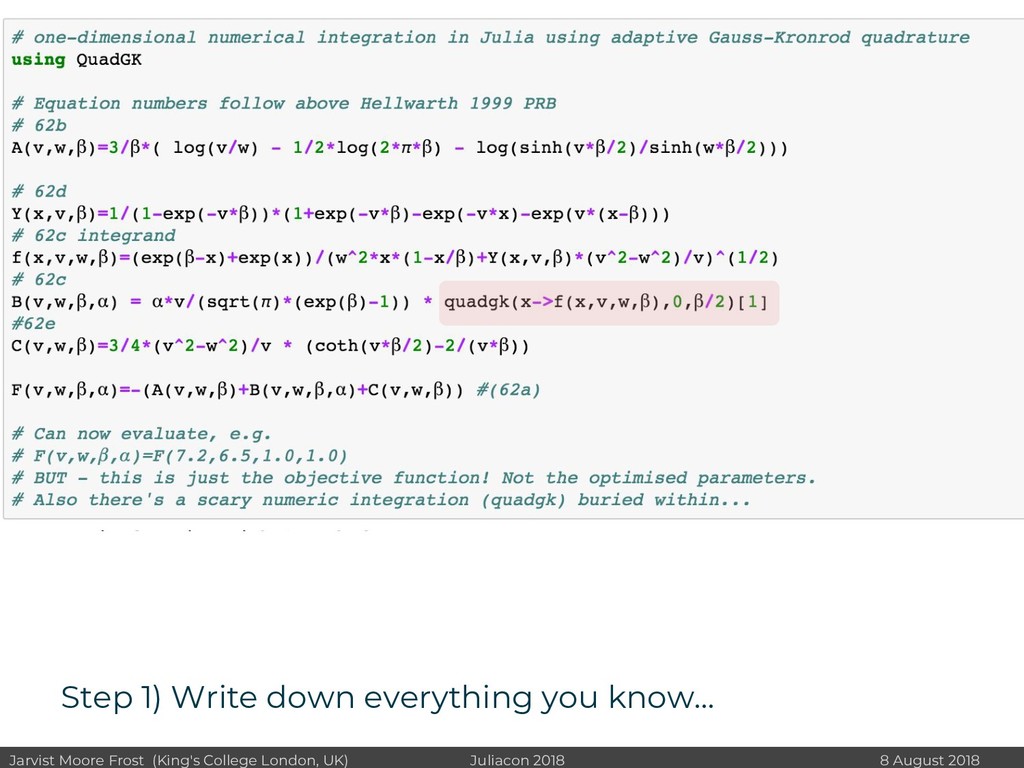
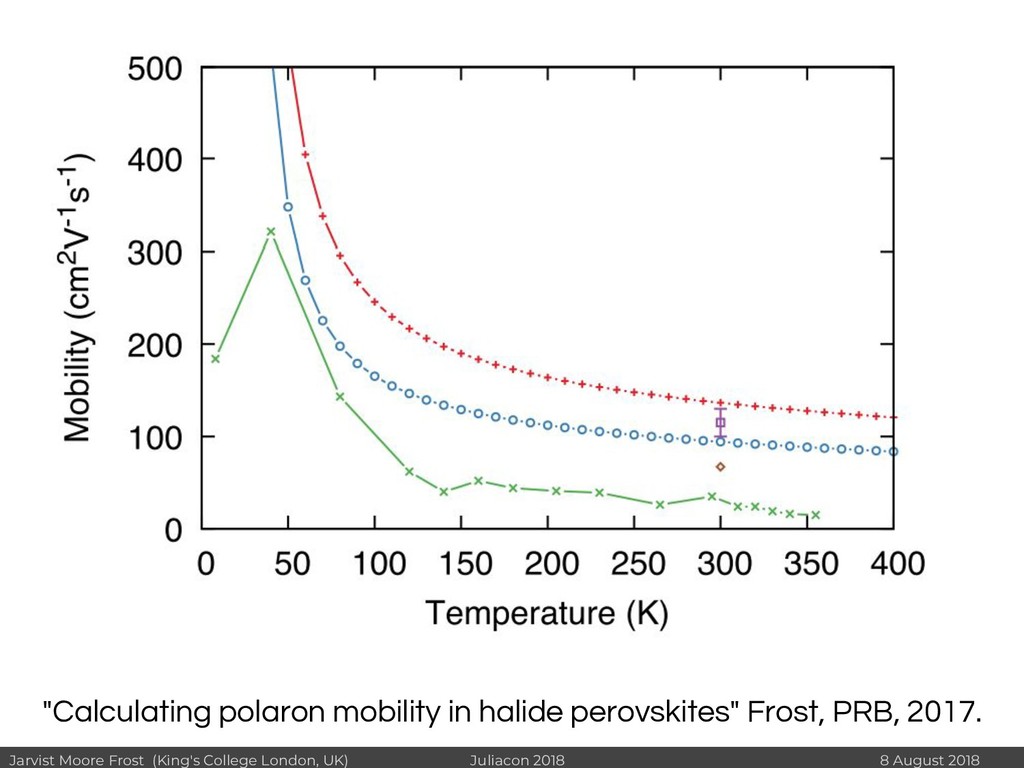
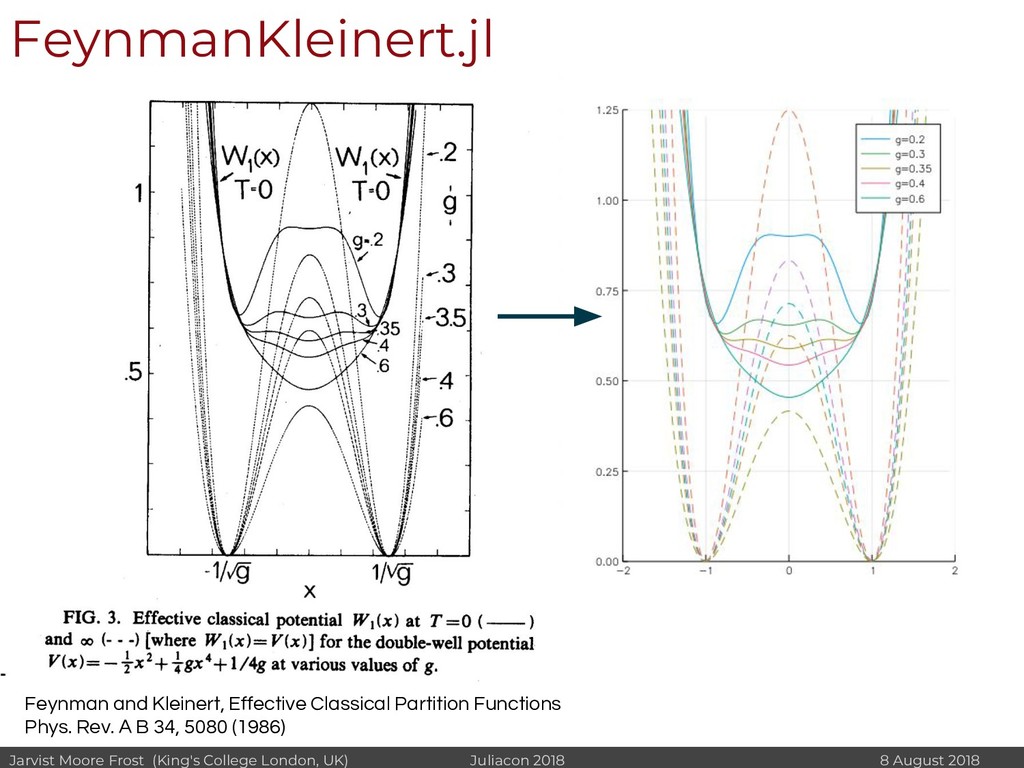
I will discuss revisiting Polaron mobility theories from the 1960s (https://github.com/jarvist/PolaronMobility.jl), approximate forms of quantum-nuclear effects (https://github.com/jarvist/FeynmanKleinert.jl) and Tight-Binding approaches to large scale electronic structure calculations.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}