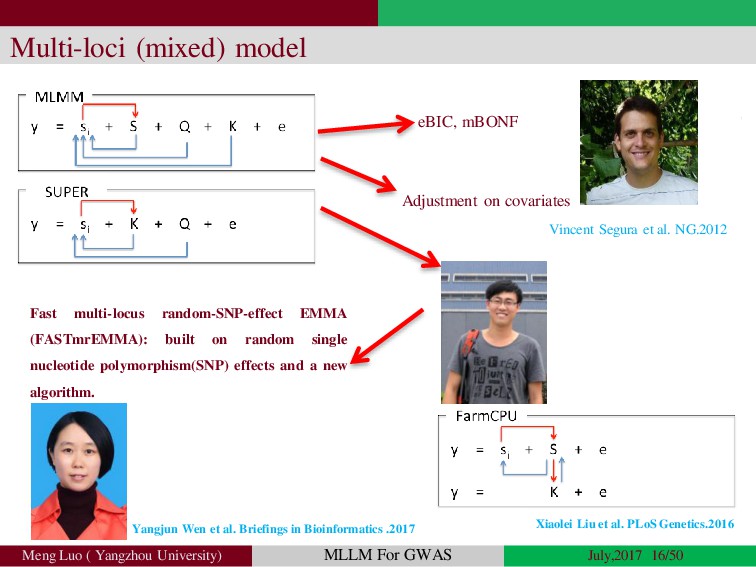
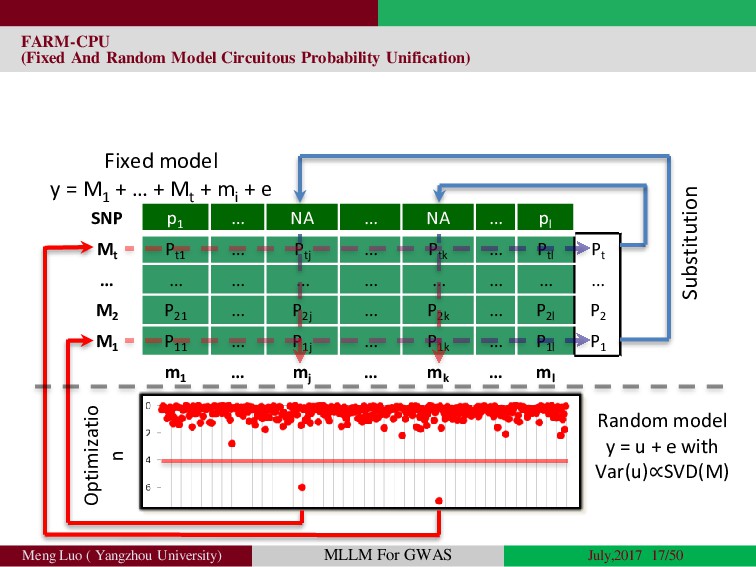
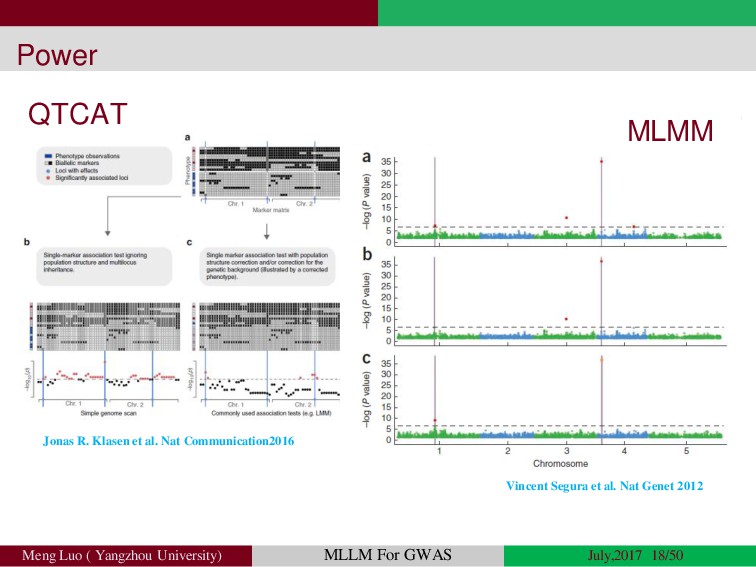
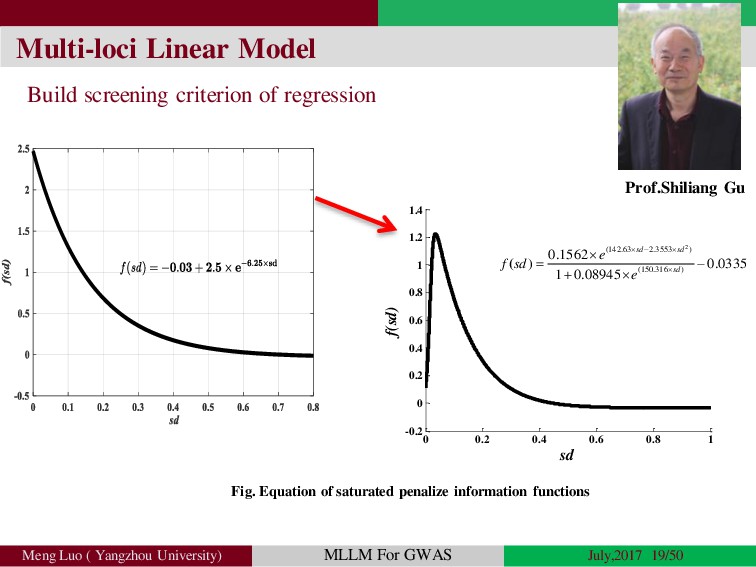
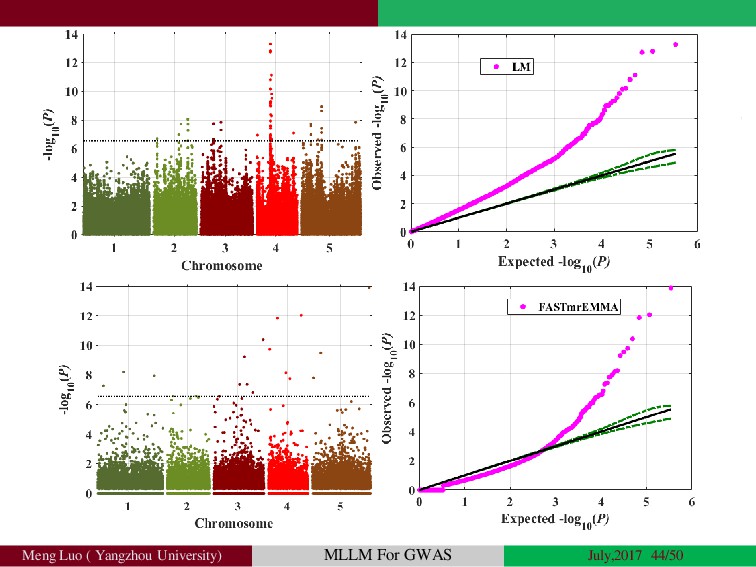
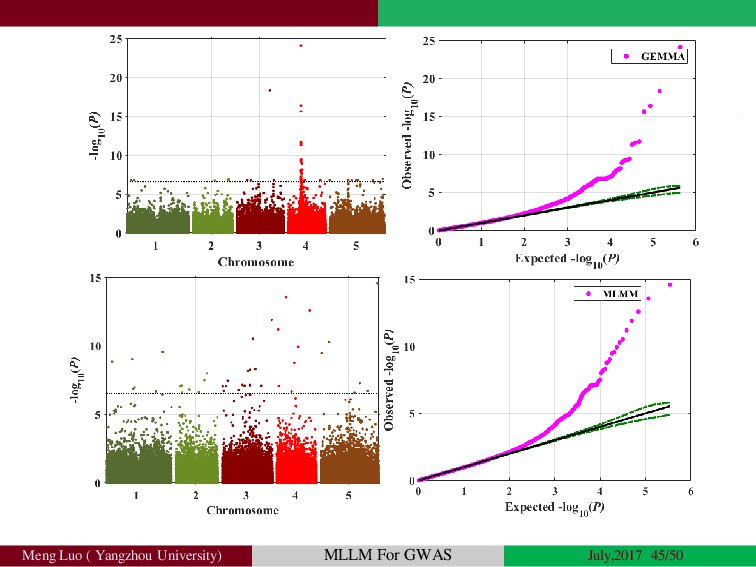
Genome-wide association studies (GWASs) have been increasingly prominent in detecting genetic variants associated with complex traits and disease, while the identified variants significant explain only a fraction of total phenotypic variance, resulting in the so-called ‘missing heritability’, but sporadically pinpointing biological mechanisms. Commonly, the individuals used in GWA studies are not related to each other, some degrees of confounding cryptic relatedness and population stratification are inevitable. During the past decades, there are many solutions to the problem of population structure, including genomic control(GC)(Devlin and Roeder, 1999; Zheng, et al., 2006), structured association (SA)(Patterson, et al., 2006; Pritchard, et al., 2000; Raj, et al., 2014), regression control (RC)(Setakis, et al., 2006; Wang, et al., 2005), principal components adjustment (PCA)(Price, et al., 2006; Zhang, et al., 2003) and mixed regression models(MRM)(Kang, et al., 2008; Price, et al., 2010; Yu, et al., 2006). Introduction
{kind=link}
{kind=link}
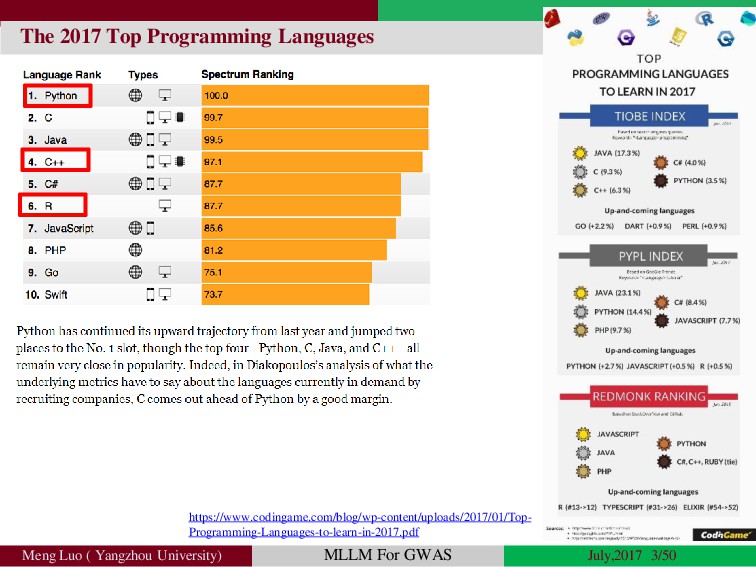{kind=link}
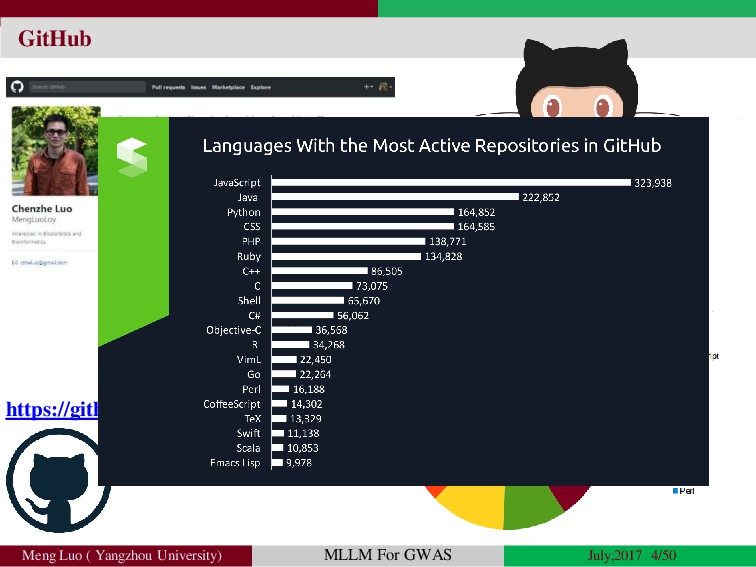{kind=link}
{kind=link}
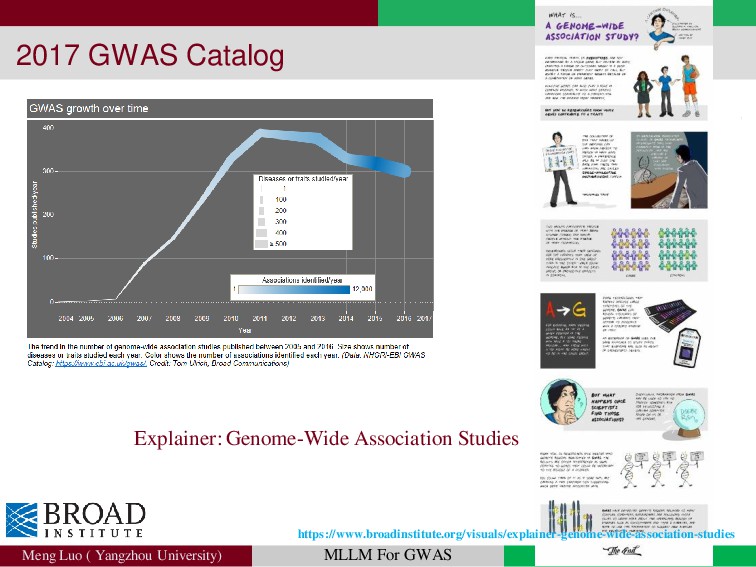{kind=link}
{kind=link}
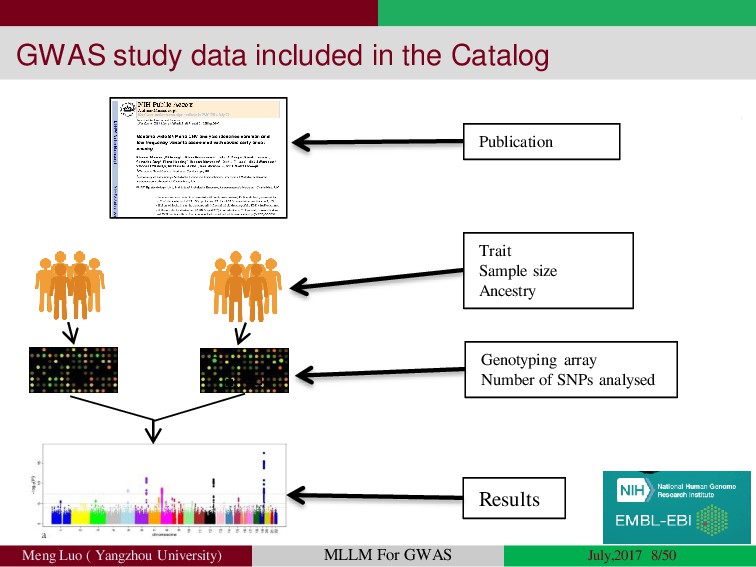{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
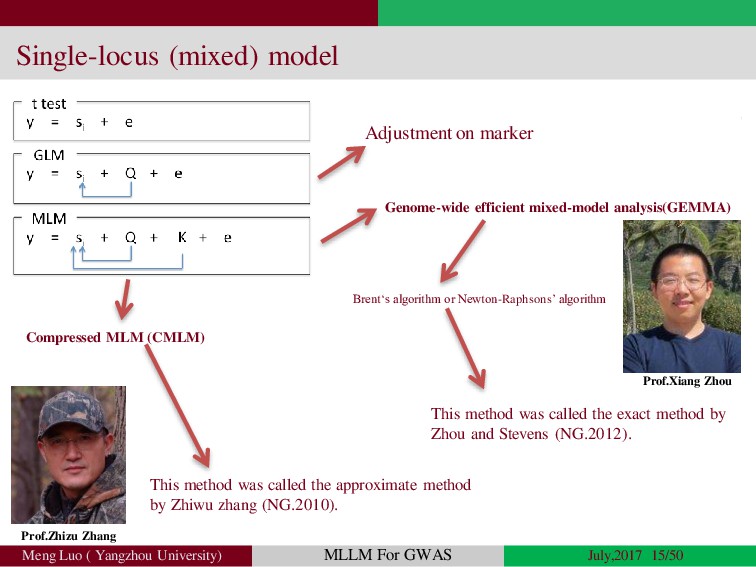{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
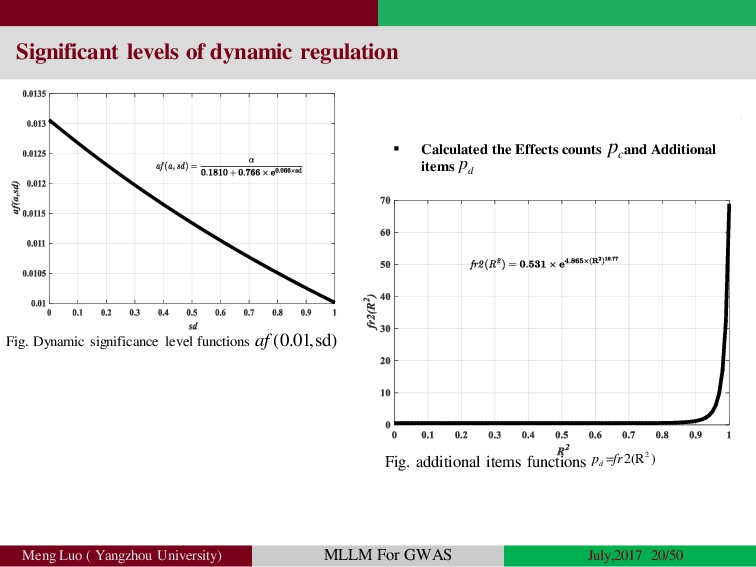{kind=link}
{kind=link}
{kind=link}
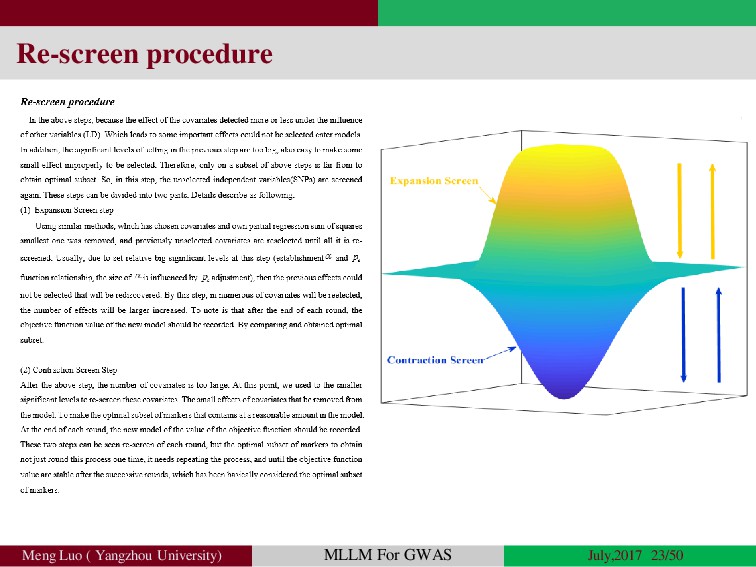{kind=link}
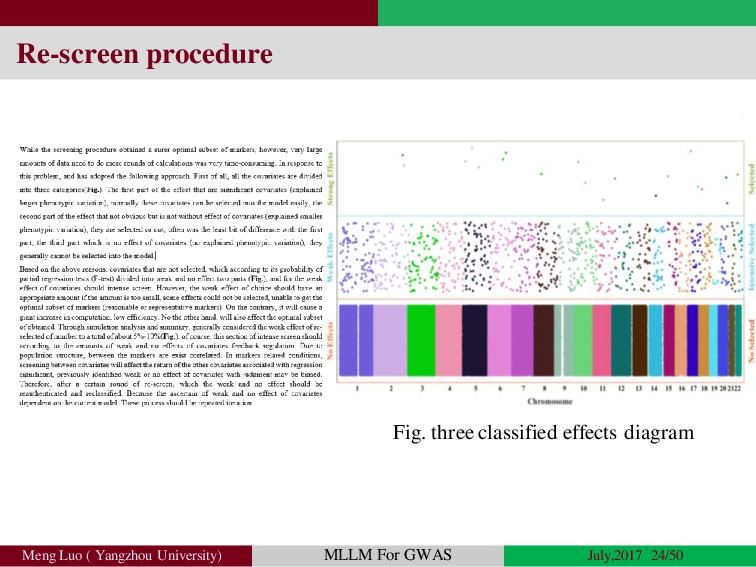{kind=link}
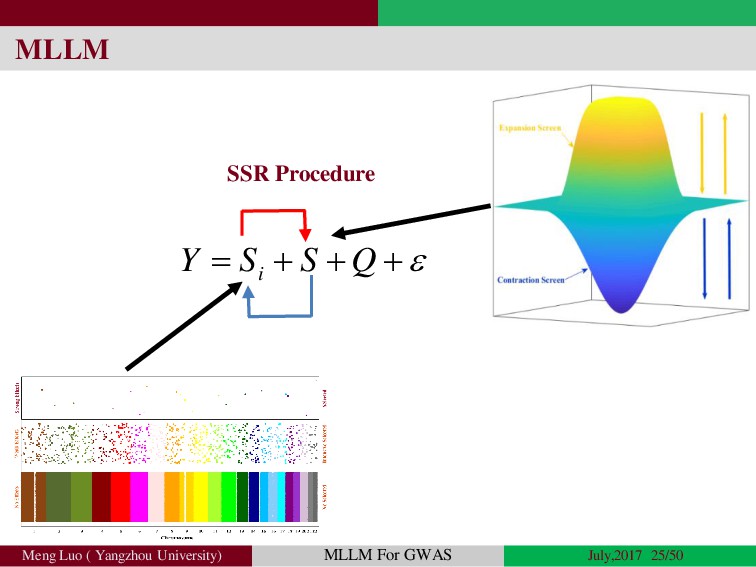{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
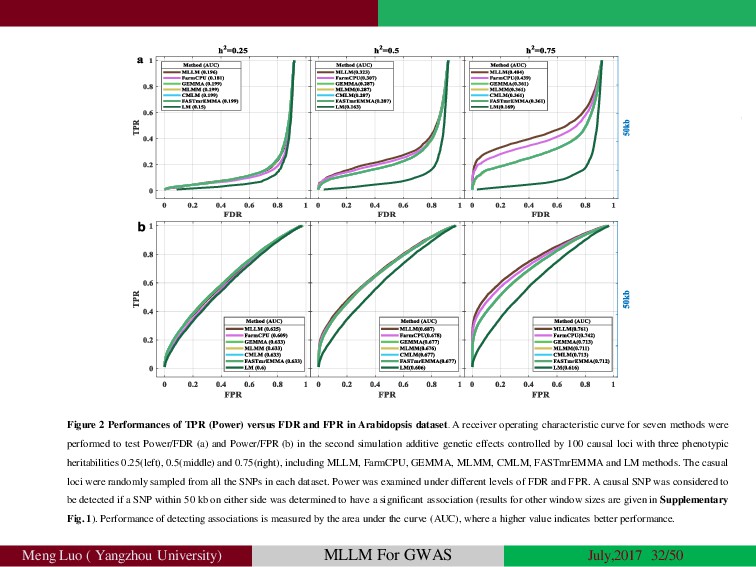{kind=link}
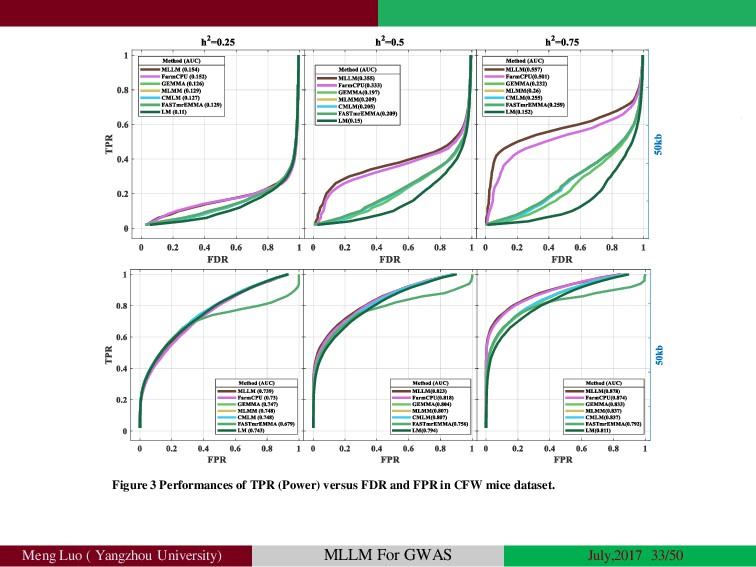{kind=link}
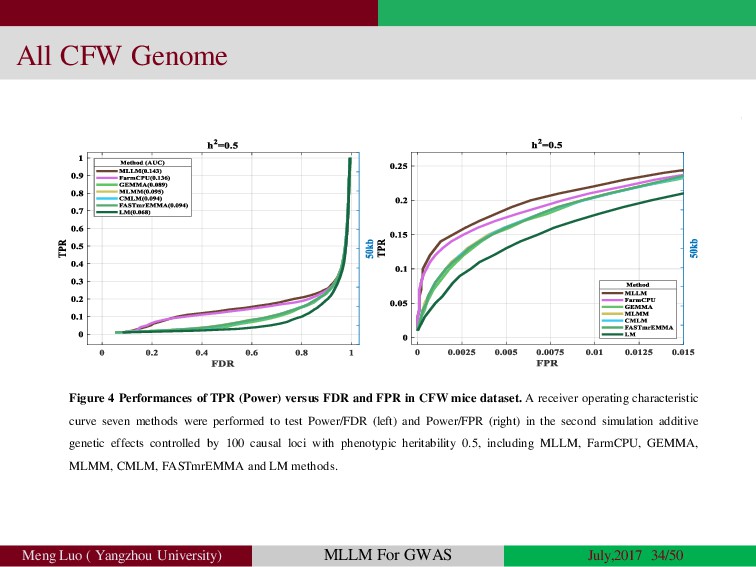{kind=link}
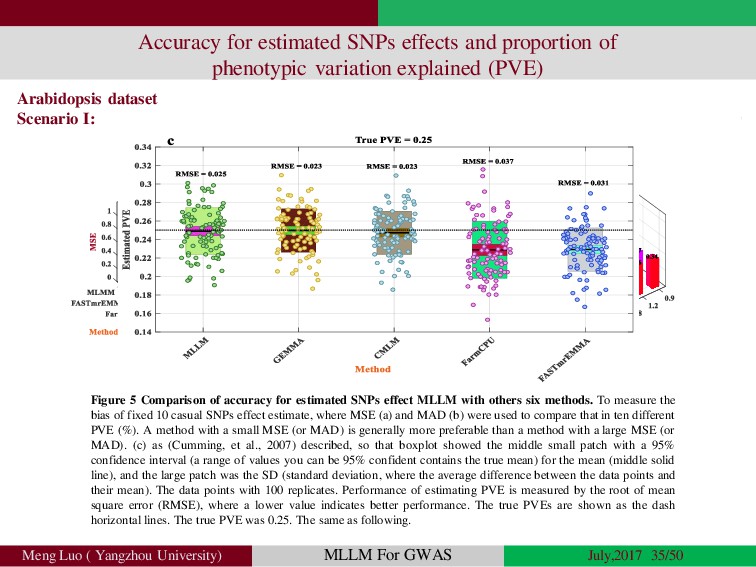{kind=link}
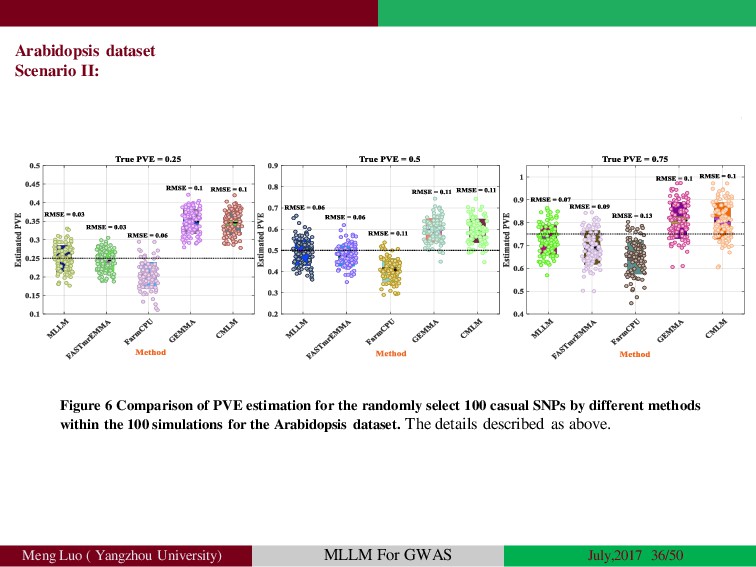{kind=link}
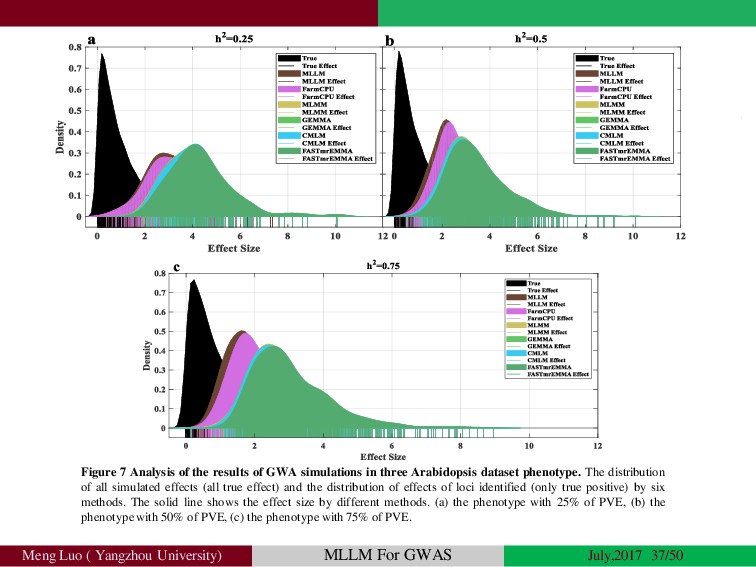{kind=link}
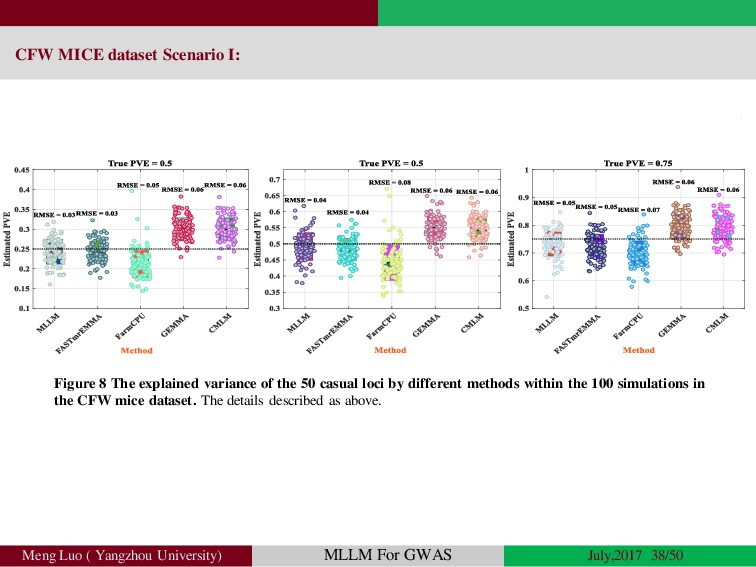{kind=link}
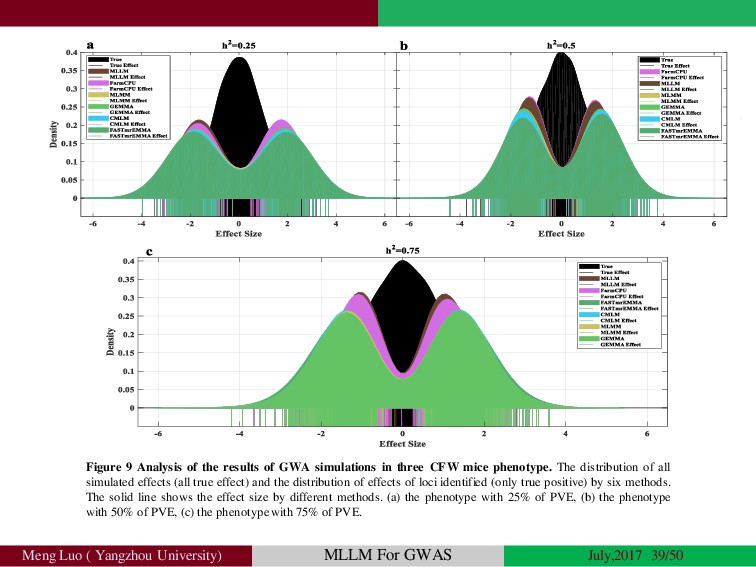{kind=link}
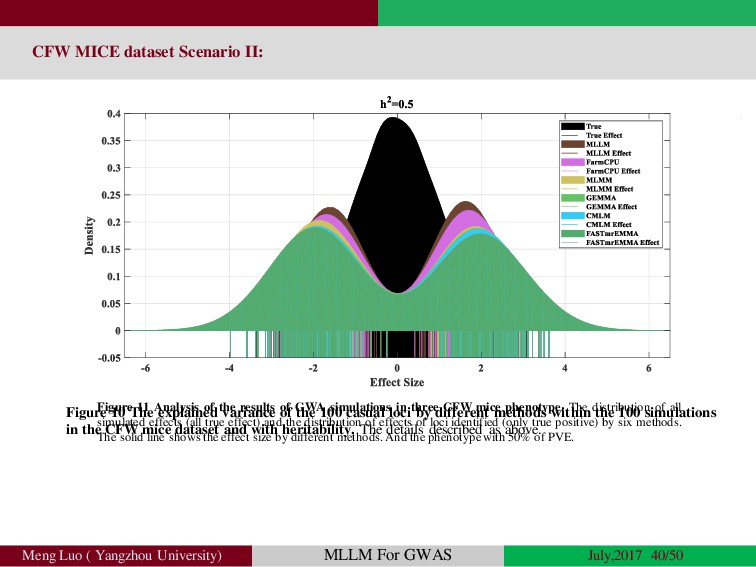{kind=link}
{kind=link}
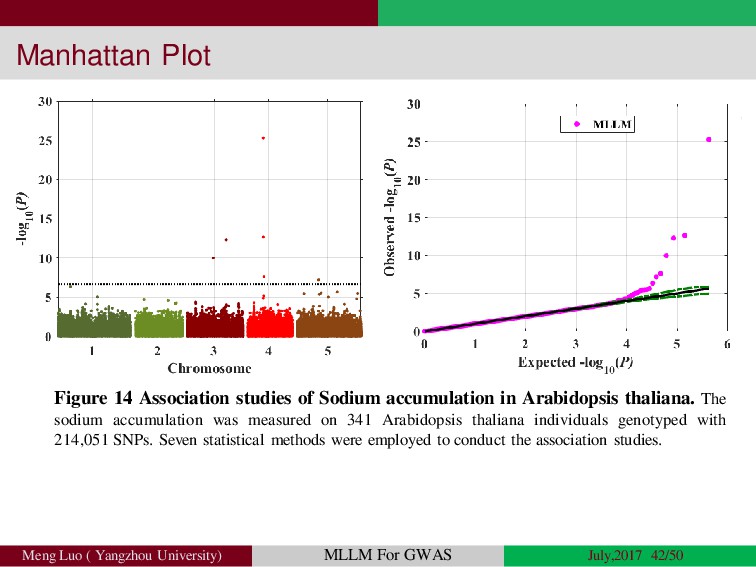{kind=link}
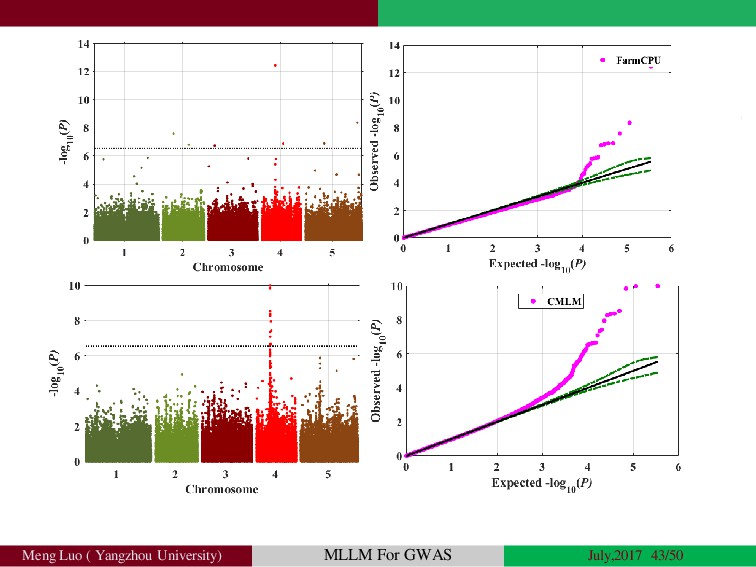{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}