for GWAS&E-GWAS&GS 攻读硕士学位期间的研究成果 • Tao Li, Meng Luo, Dadong Zhang, Di Wu, Lei Li, Guihua Bai. Effective marker alleles associated with type 2 resistance of wheat to Fusarium head blight infection in fields. Breeding Science, 2016, 66(3):350-357. • 钱丹,骆孟,董晶晶,李长成,李磊,李韬.小麦品种宁7840突变体农艺性状和赤霉病抗性解析[J]. 麦类作物学报 . 2016(02) 119 • 李韬,骆孟,钱丹,董晶晶,顾世梁.抗赤霉病小麦地方品种黄方柱和海盐种EMS突变体的变异分析[J]. 植物遗传资源学 报 . 2016(06) 71 • 龚璇,骆孟,肖天晶,曹静,李磊,李韬.小麦突变体群体赤霉病抗性及农艺性状变异解析[J]. 扬州大学学报(农业与生命科 学版) . 2017(04) 28 • 施璇,李磊,郑彤,骆孟,李韬.小麦类过敏反应突变体对氮素的响应及对白粉病的抗性[J]. 麦类作物学报 . 2018(04) • Meng Luo, Tao Li, Shiliang Gu. An Efficient Iterative Screen Regression Method for Genome-Wide Association Studies in Structured Populations. 2018, (Current preprint (BioRxiv)). • Meng Luo, Shiliang Gu. Iterative Screen Regression Models for Genetic Mapping Studies of Epistasis of Quantitative Traits. 2018, (Current preprint (BioRxiv)). • Meng Luo, Shiliang Gu. Genetic Prediction of Complex Traits with Iterative Screen Regression Models. 2018, (Current preprint (BioRxiv)). • Meng Luo, Shiliang Gu. Solve traveling salesman problem using EMF-CE algorithm. Transactions on Evolutionary Computation. (under review). • LUO Meng, ZHANG Ming-Yan, PENG Yong-Xin, GUO Wen-Shan, ZHU Xin-Gai, LI Chun-Yan, BAI Gui-Hua, LI Tao and FENG Chao-Nian. Genome-Wide Association Analysis between SNP Markers and Zinc Content in Wheat Grains. Acta Agronomica Sinica, (in Chinese and under review). Core Journals! Unpublished
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
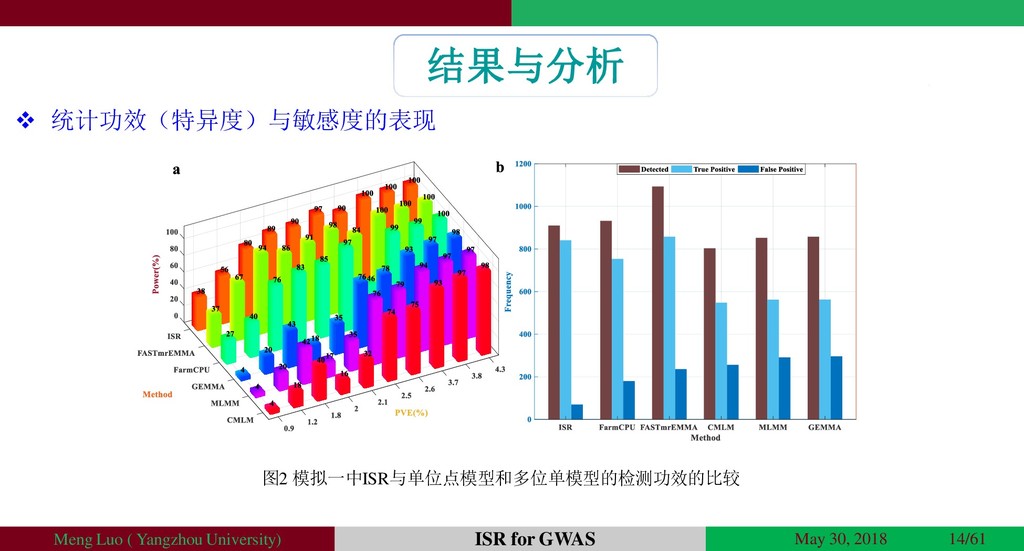{kind=link}
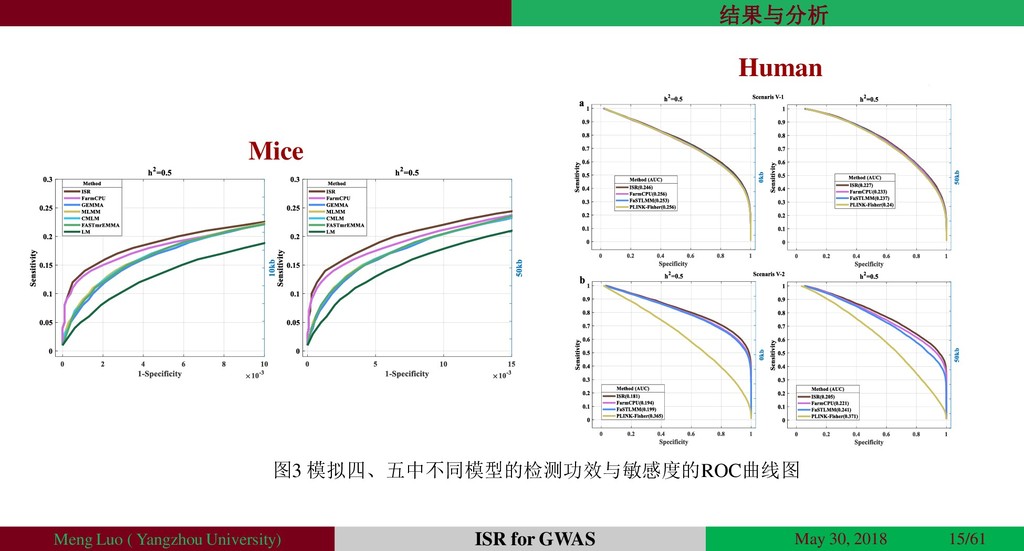{kind=link}
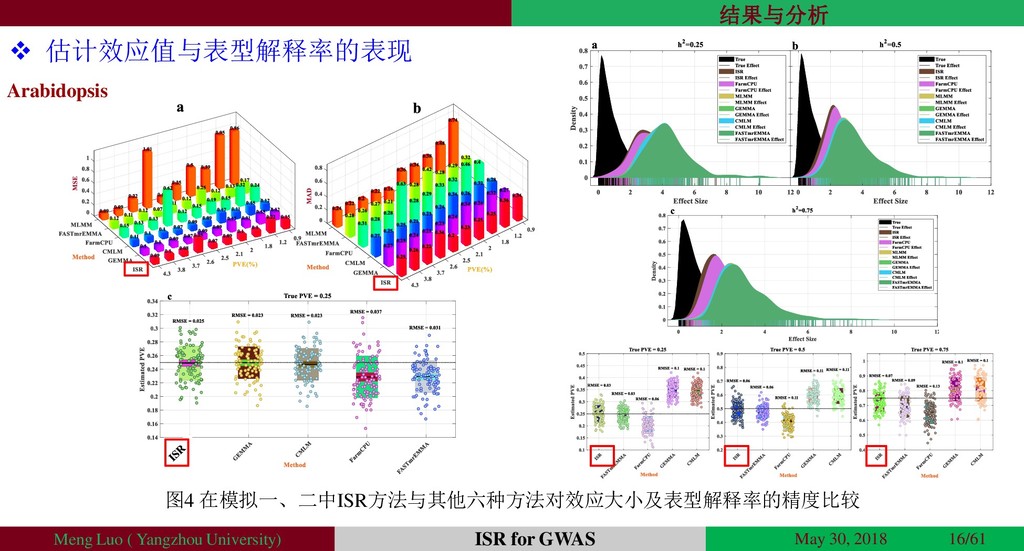{kind=link}
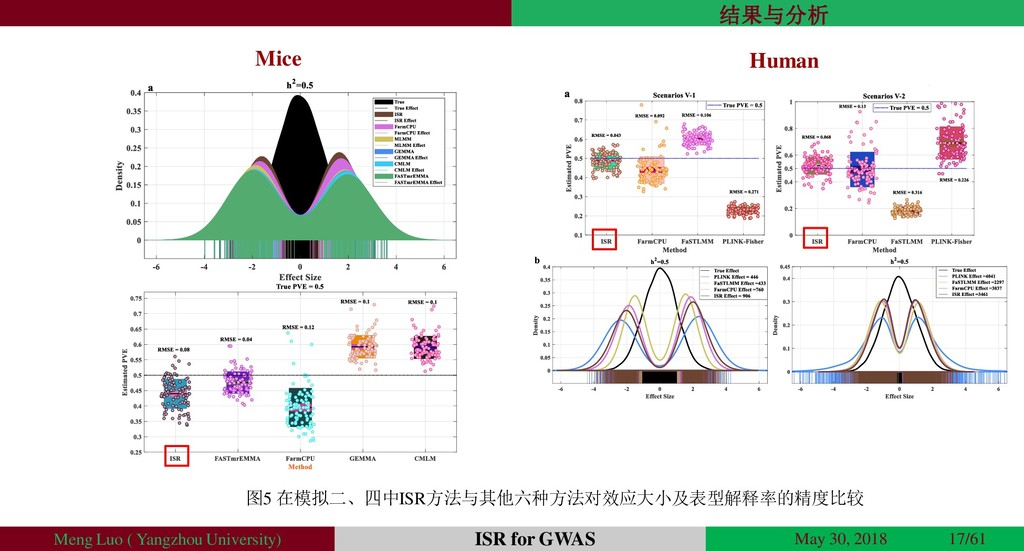{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
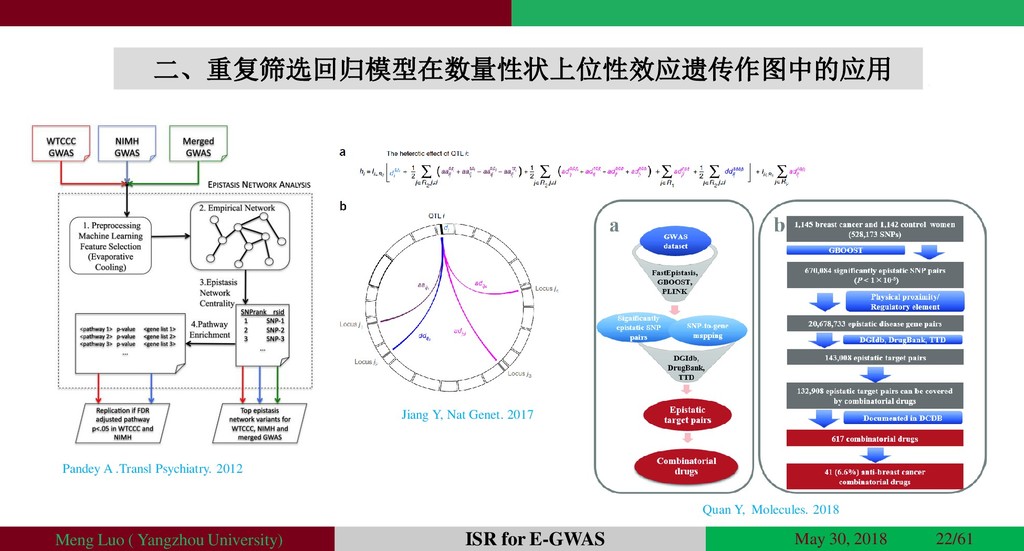{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
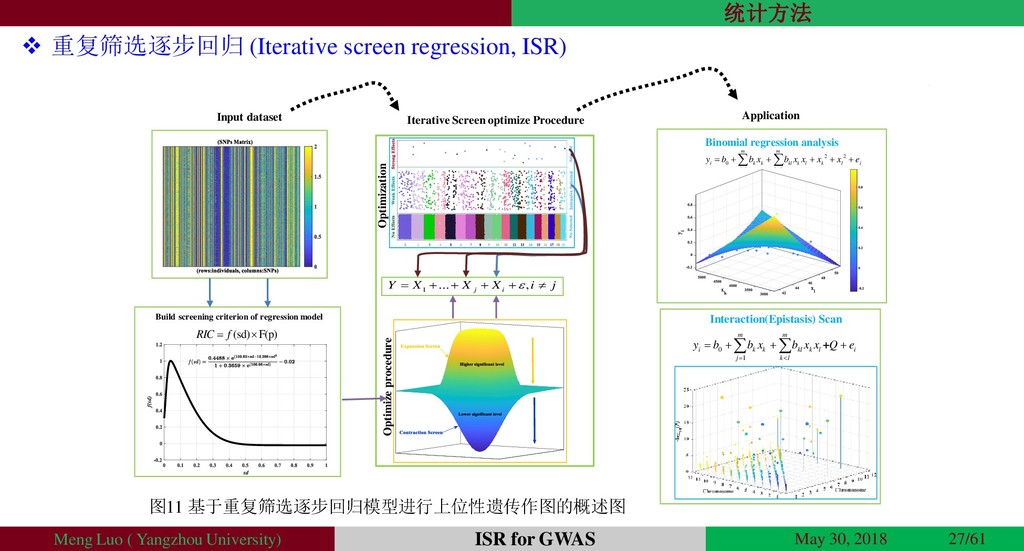{kind=link}
{kind=link}
{kind=link}
{kind=link}
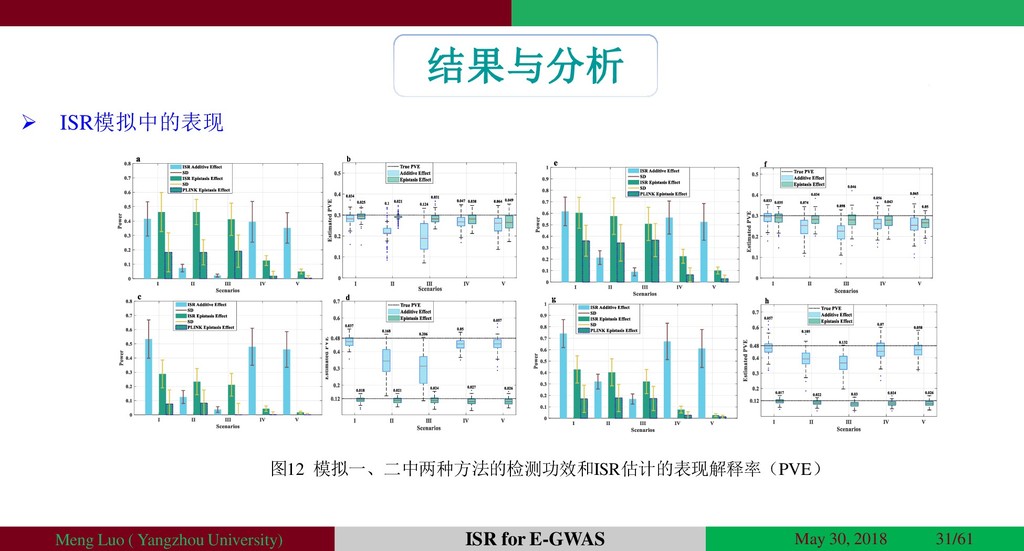{kind=link}
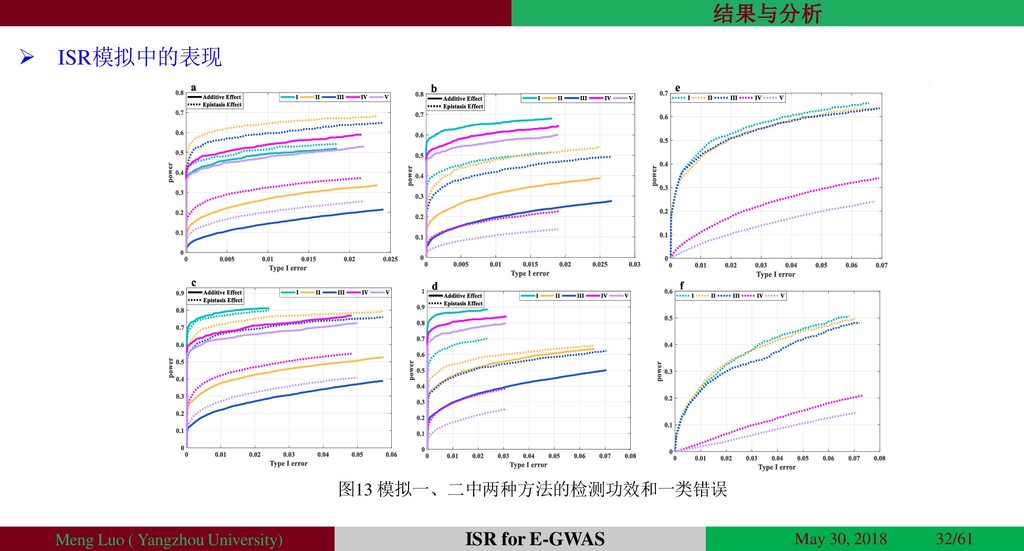{kind=link}
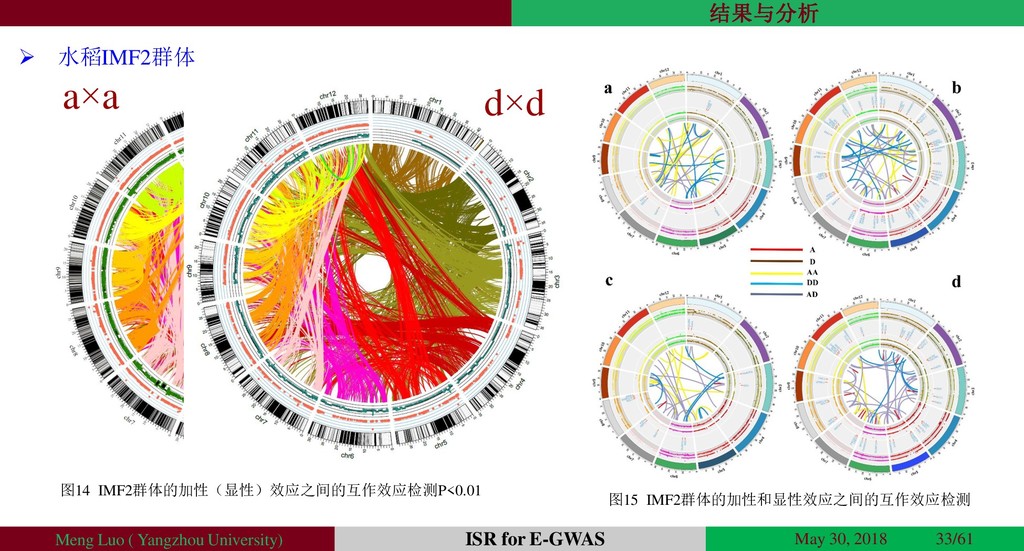{kind=link}
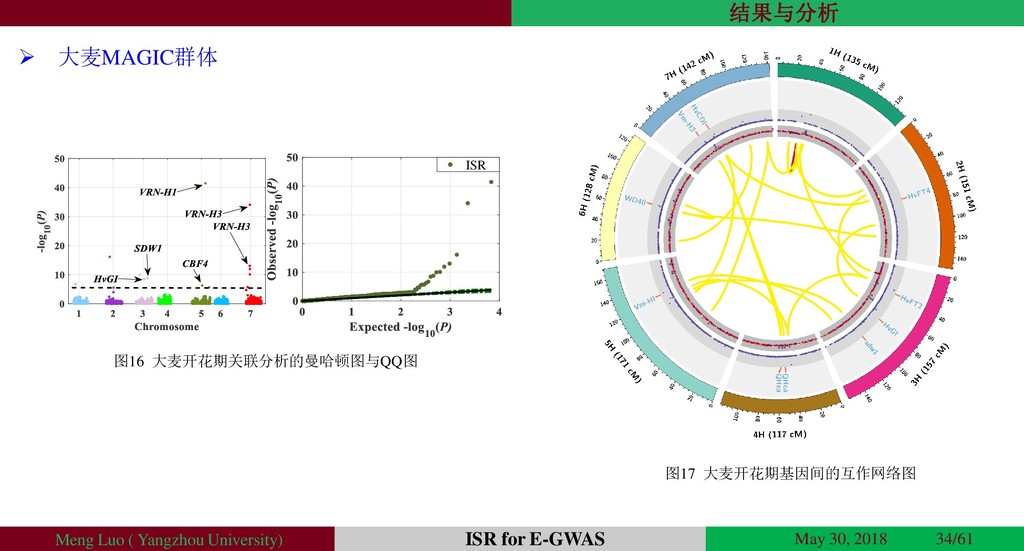{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
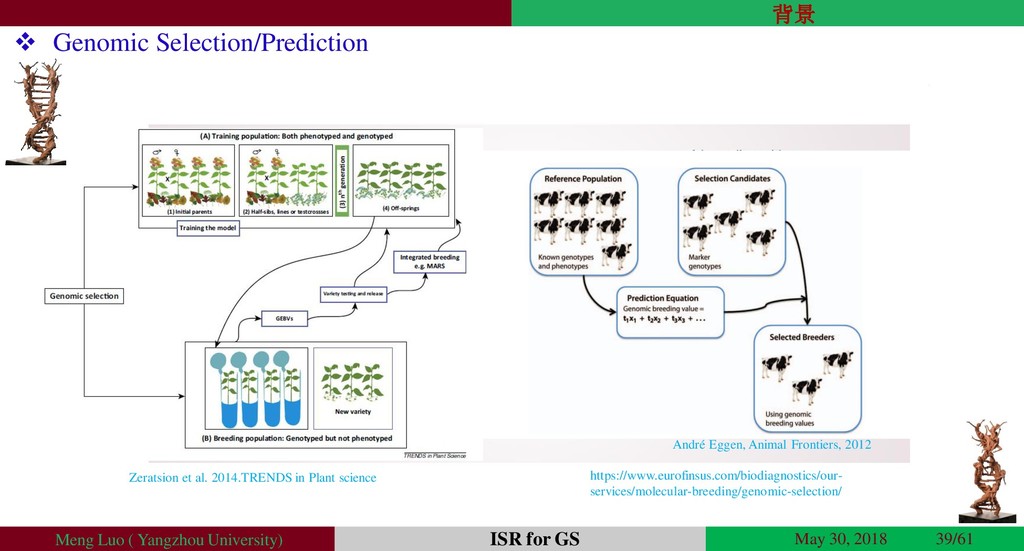{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
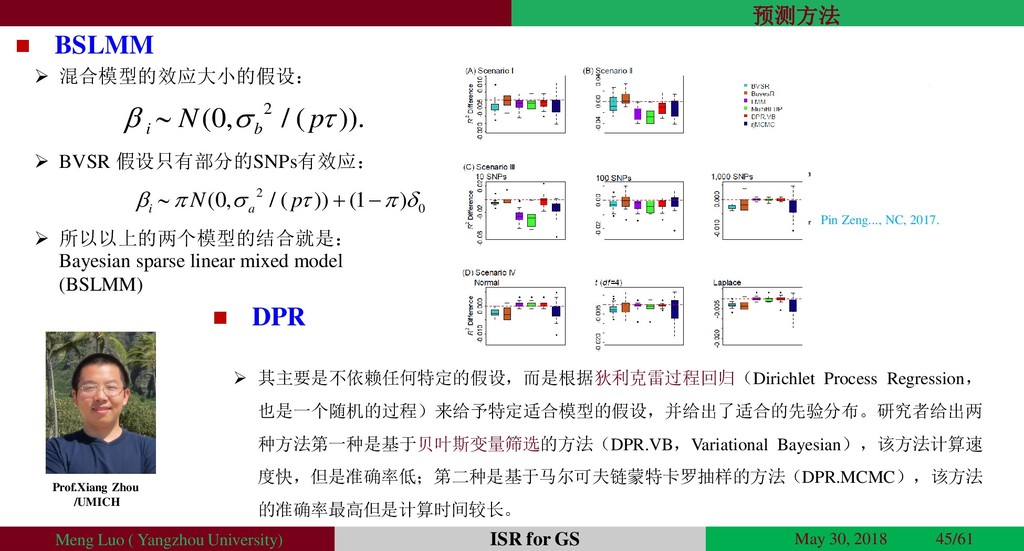{kind=link}
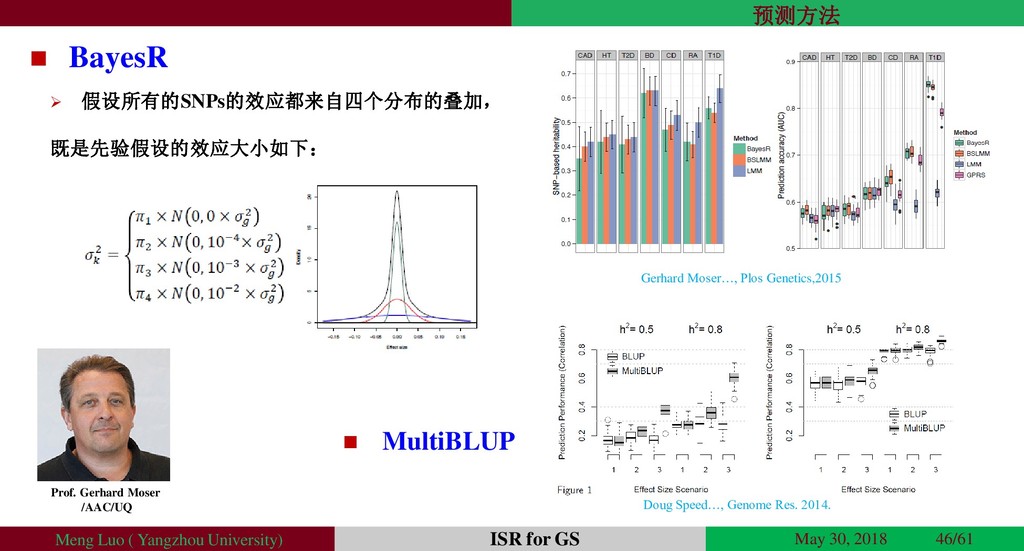{kind=link}
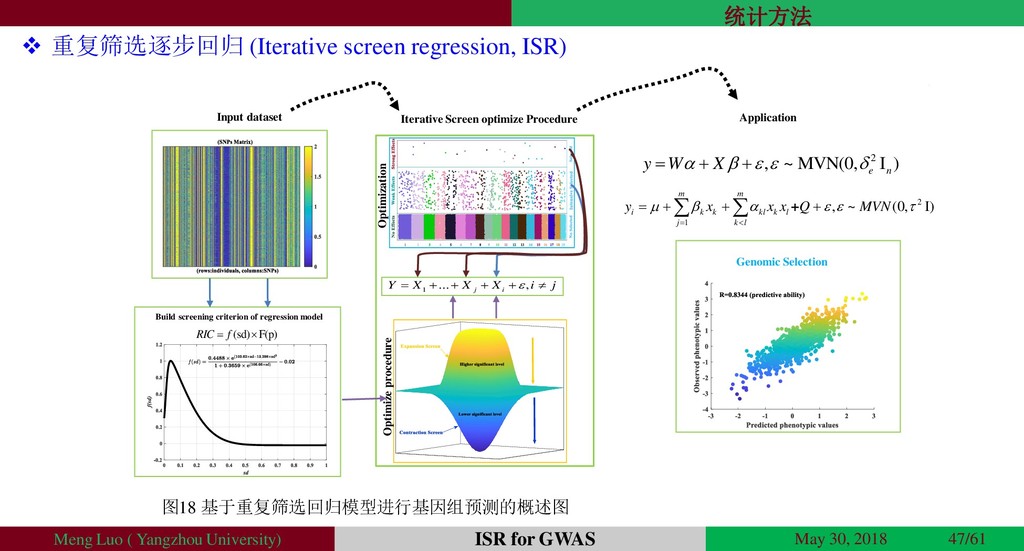{kind=link}
{kind=link}
{kind=link}
{kind=link}
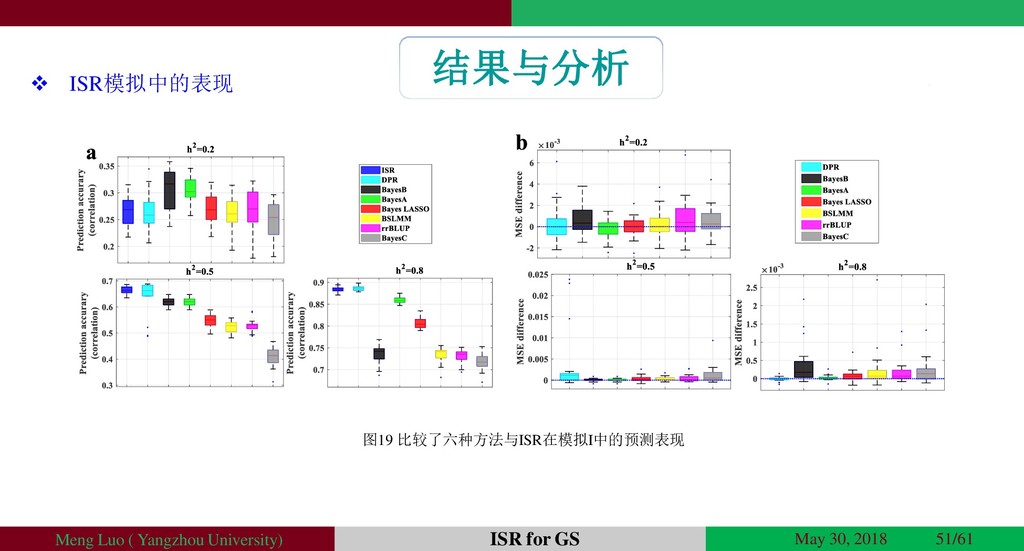{kind=link}
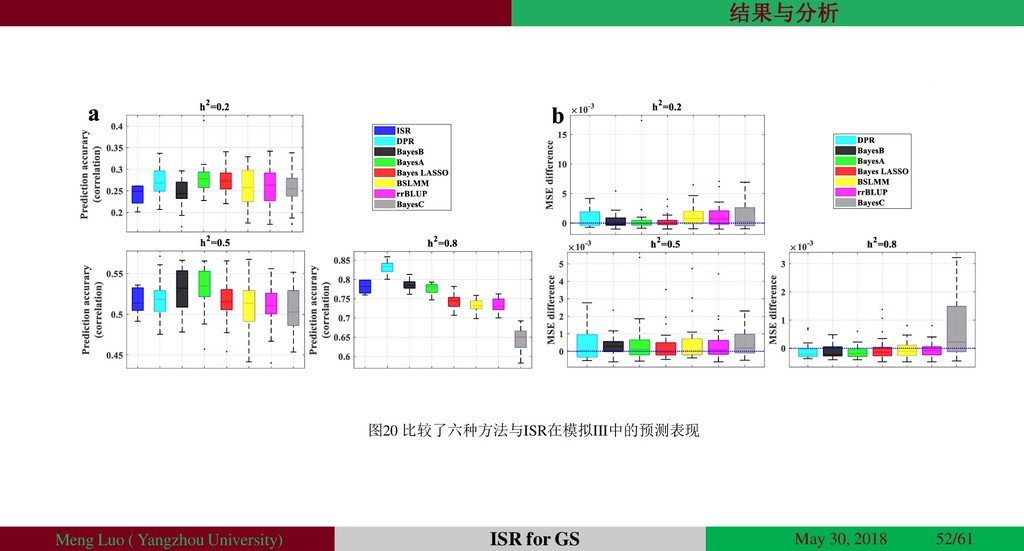{kind=link}
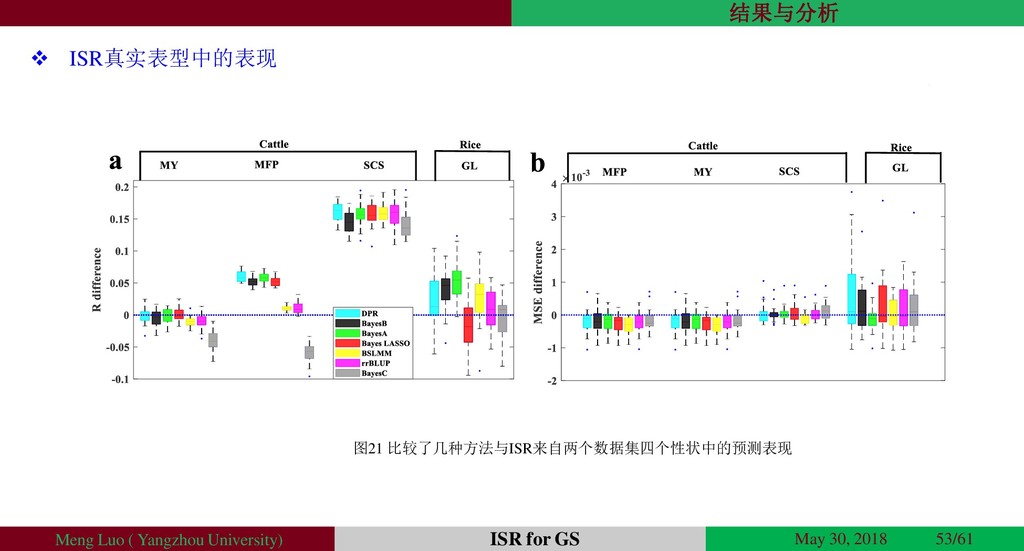{kind=link}
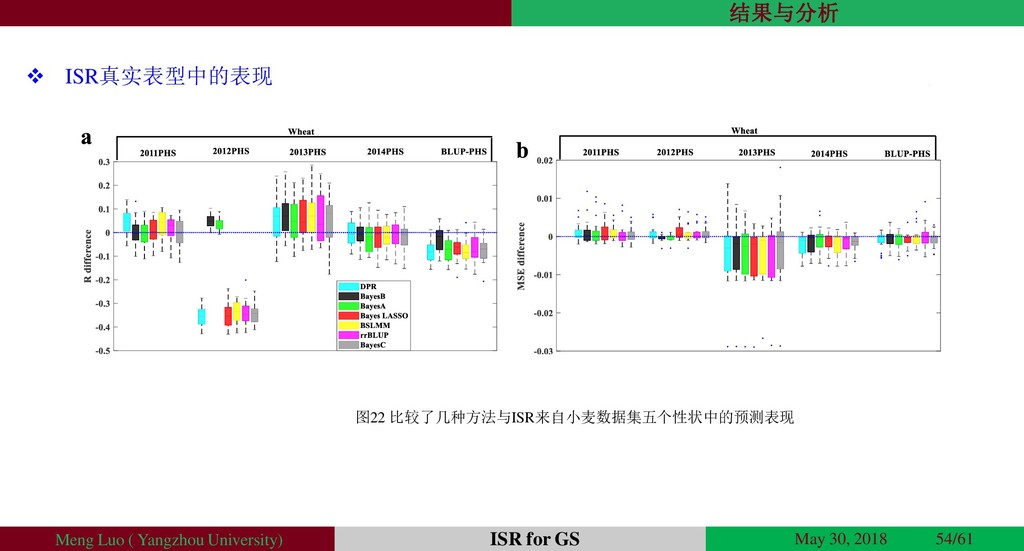{kind=link}
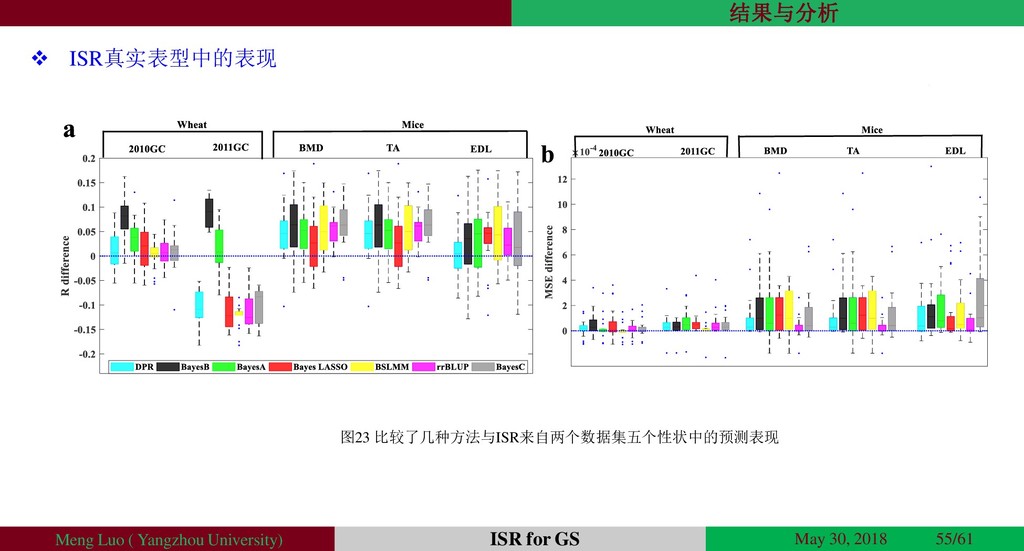{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}