@stevemunger Post-‐doctoral Fellow Gary Churchill Group The Jackson Laboratory Bar Harbor, Maine USA Duke Molecular Physiology InsNtute NGS Interest Group Slides are posted on www.speakerdeck.com/stevemunger
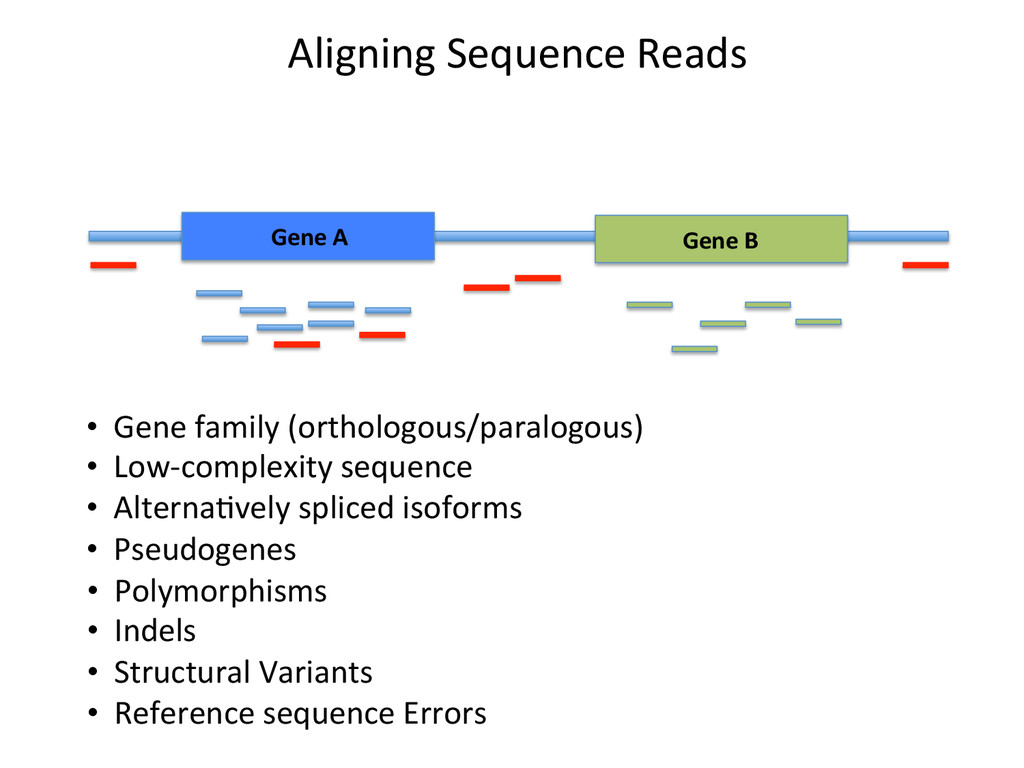
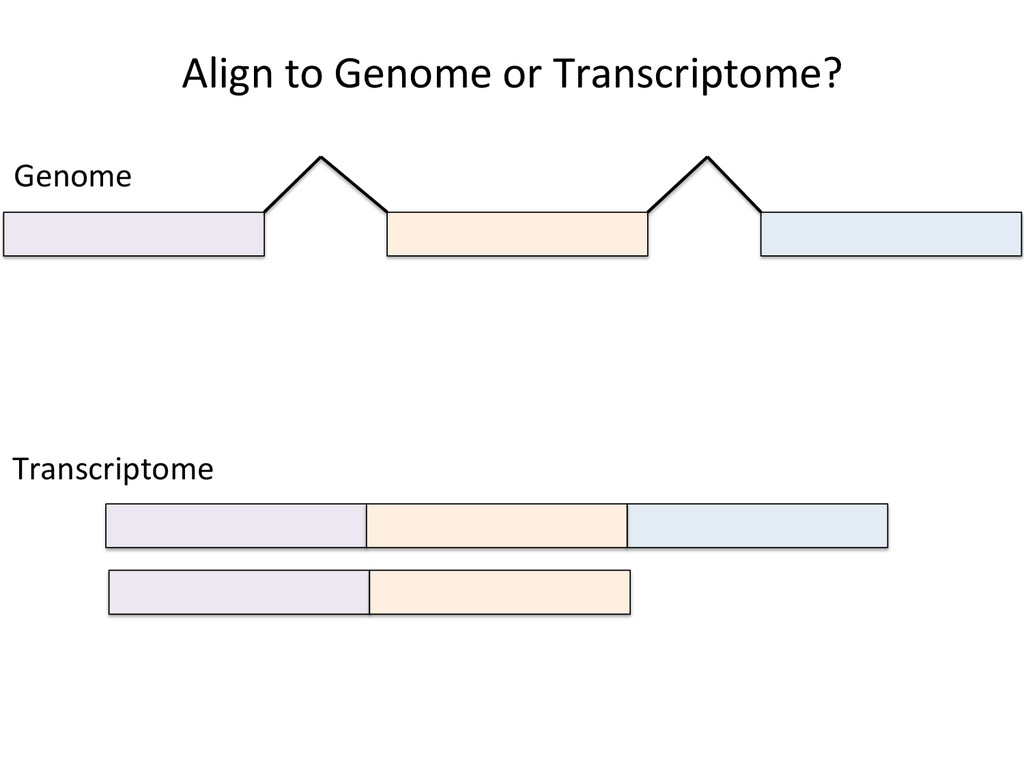
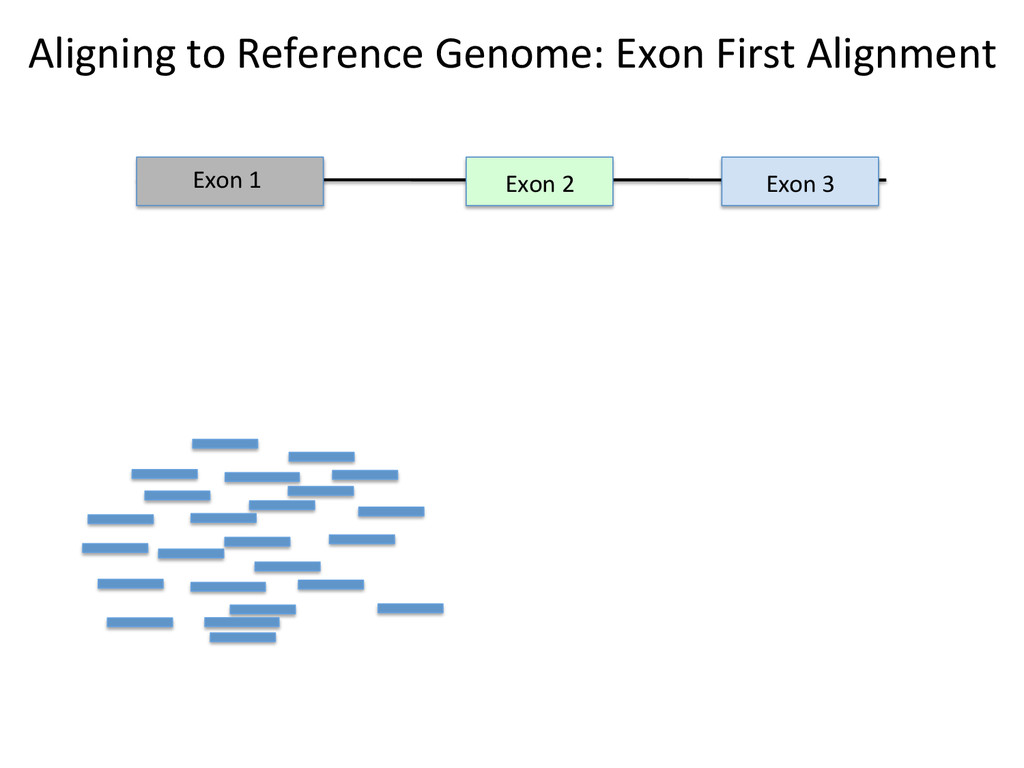
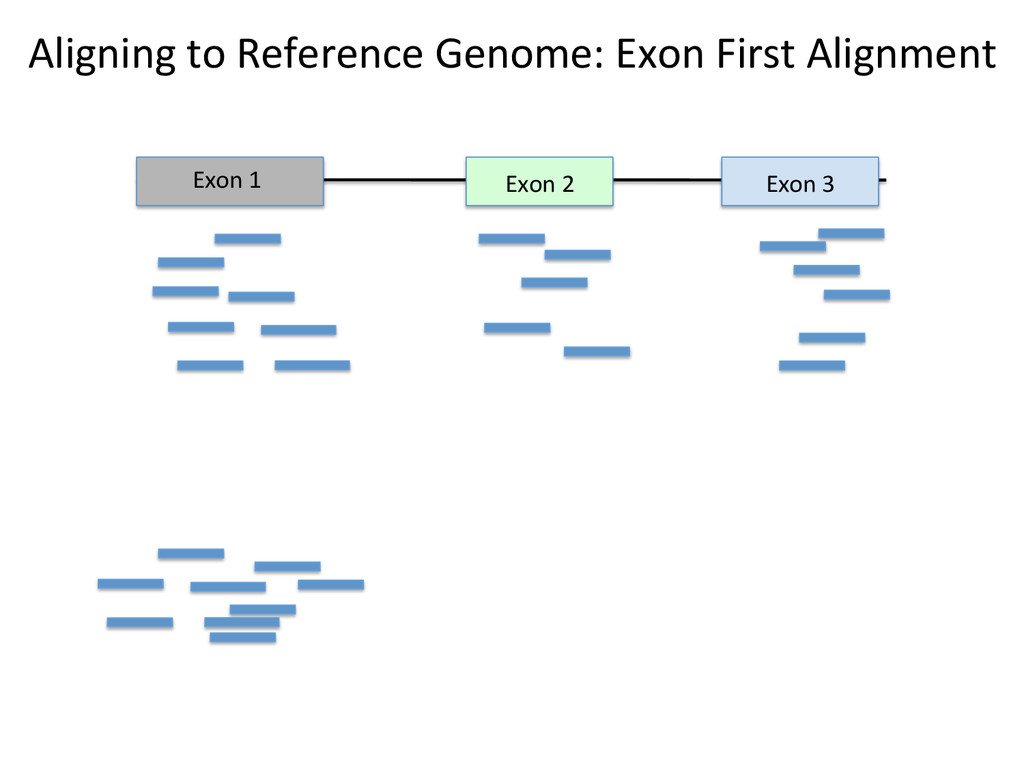
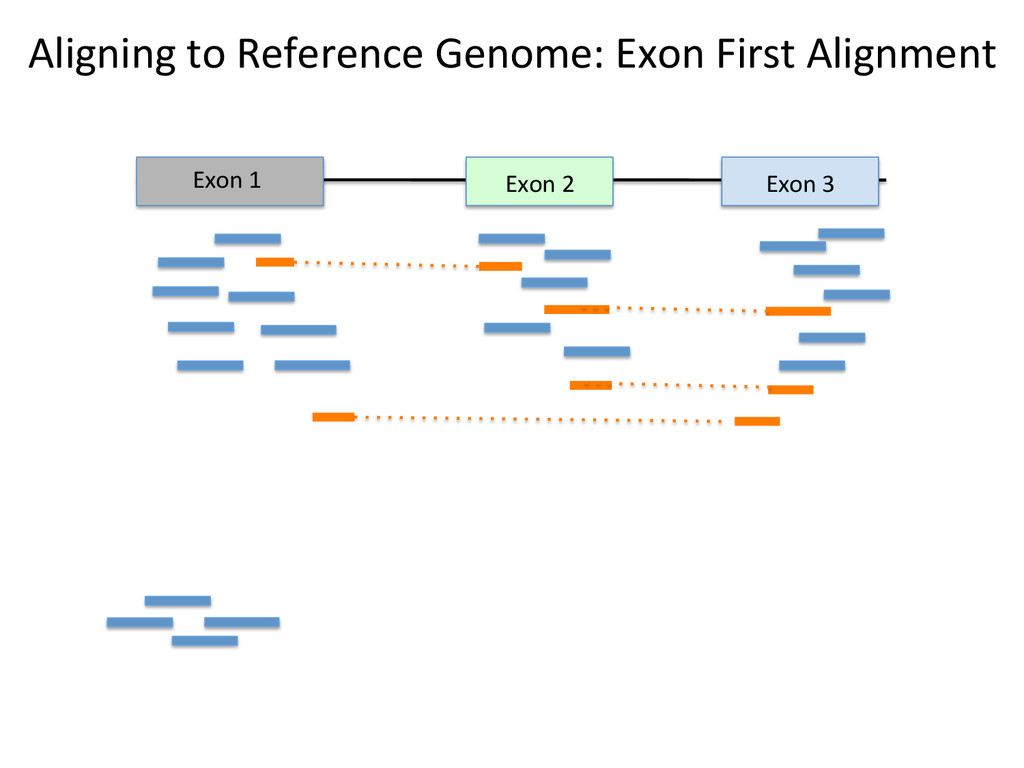
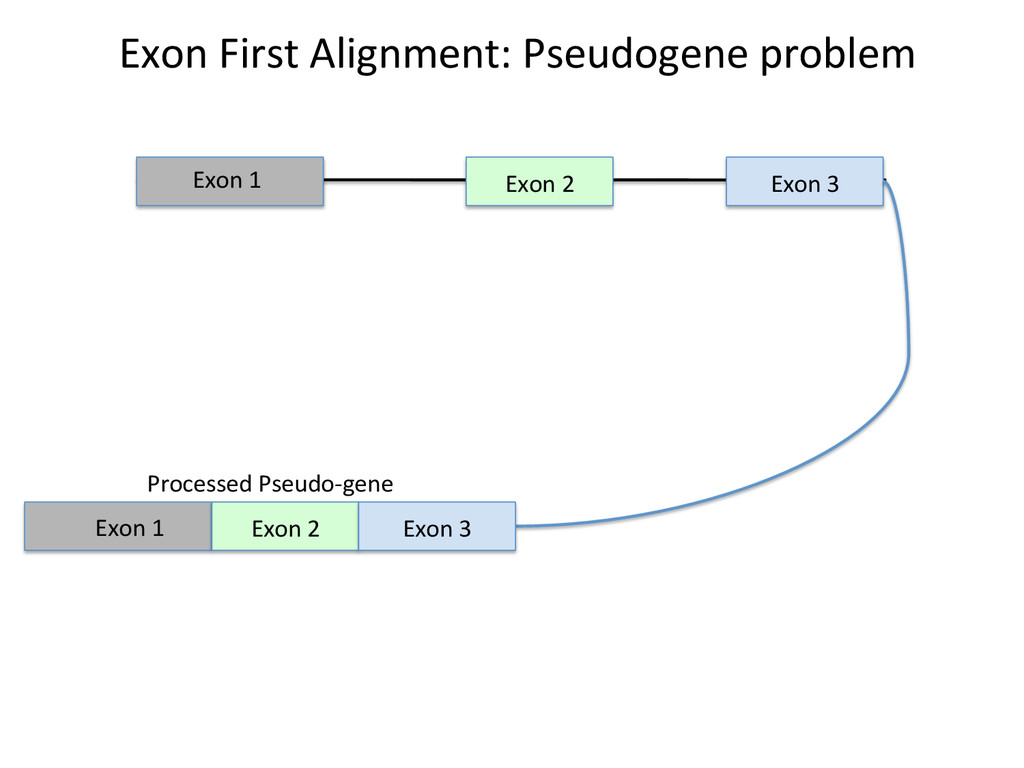
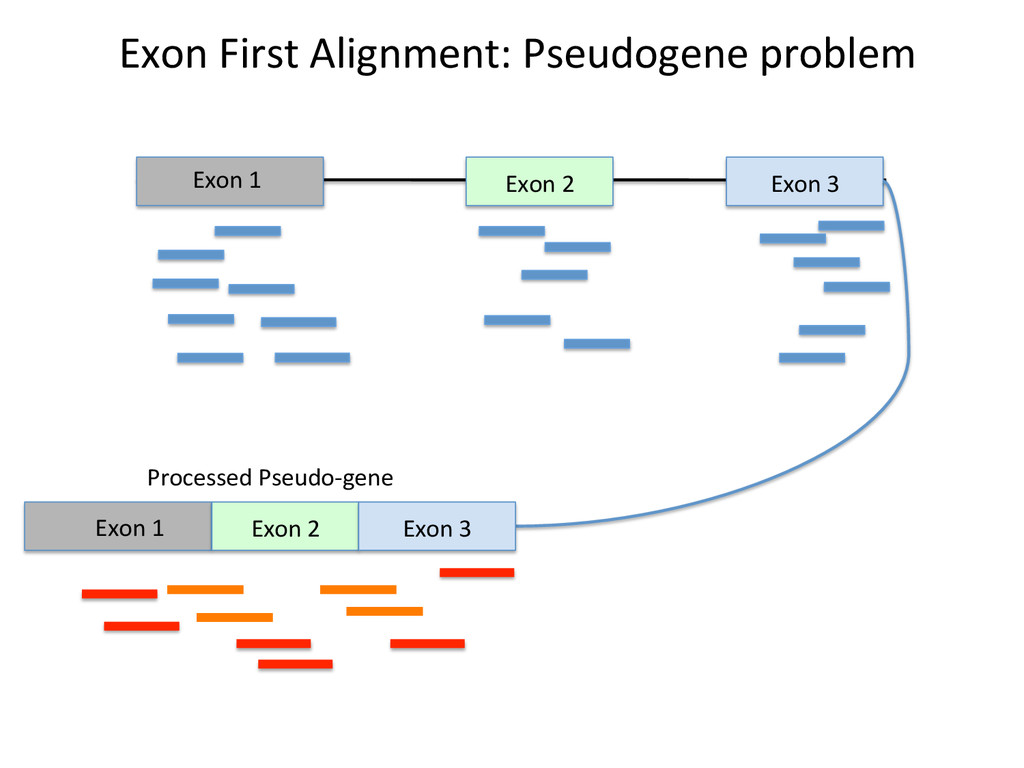
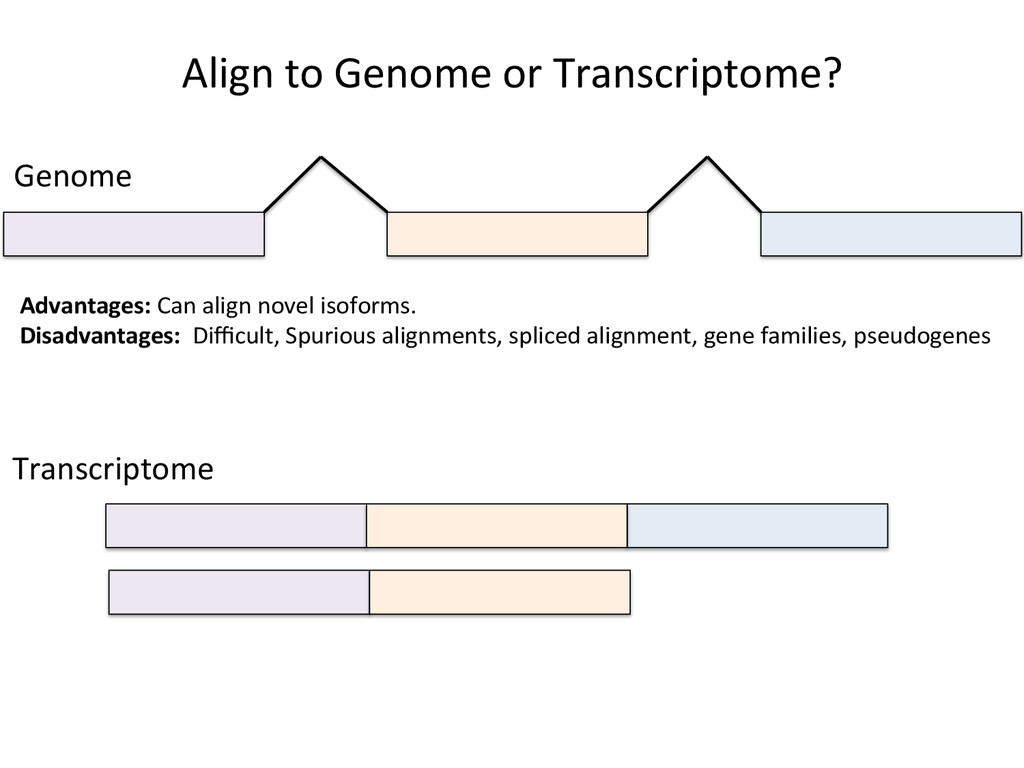
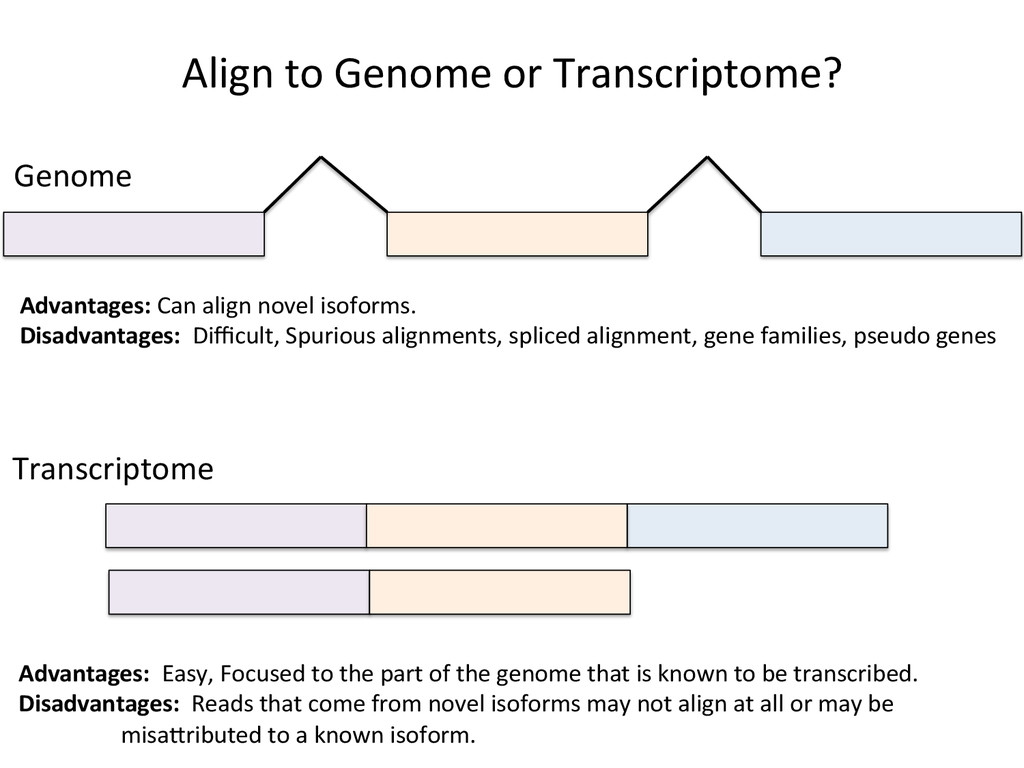
Advantages: Easy, Focused to the part of the genome that is known to be transcribed. Disadvantages: Reads that come from novel isoforms may not align at all or may be misafributed to a known isoform. Advantages: Can align novel isoforms. Disadvantages: Difficult, Spurious alignments, spliced alignment, gene families, pseudo genes
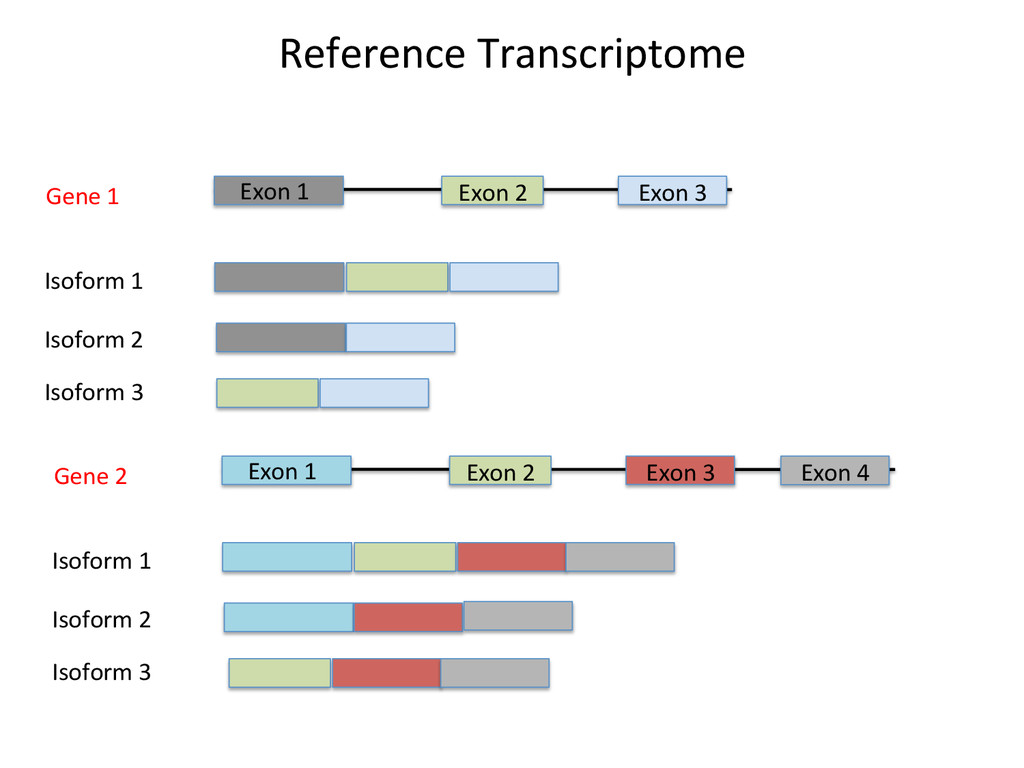
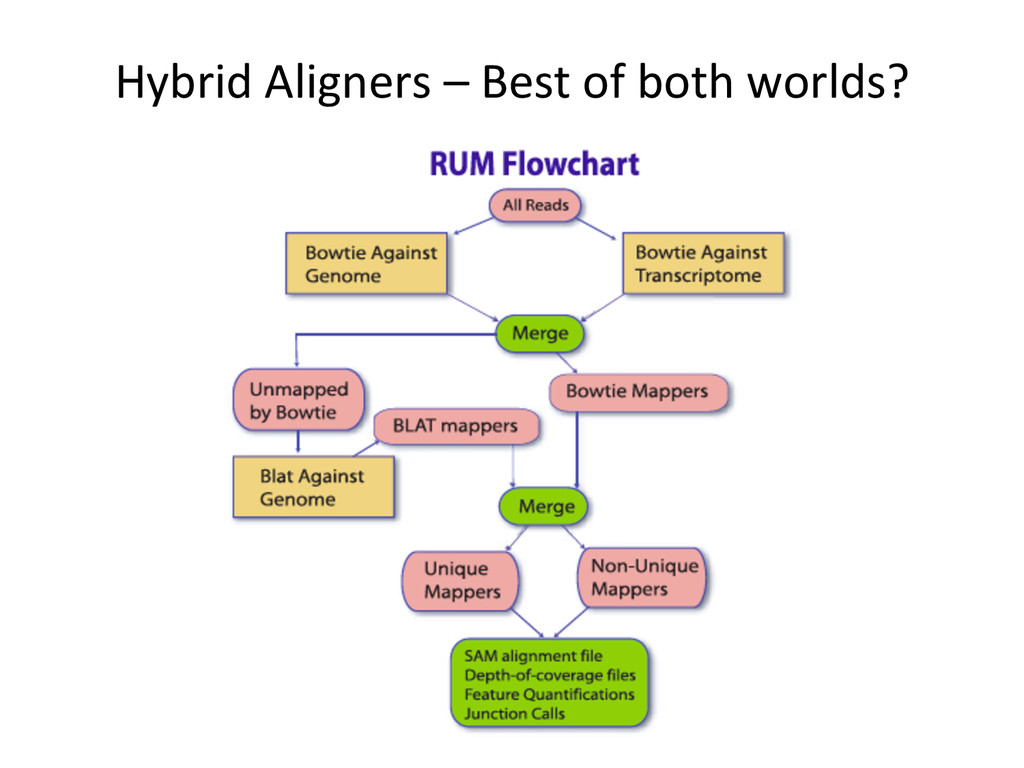
Transcriptome First Advantages: relaNvely simpler, overcomes the pseudo-‐gene and novel isoform problems Align the remaining reads to Genome next RUM, TopHat2, STAR
well-‐suited to your applicaNon. – E.g. Want to idenNfy novel exons? Don’t align only to the known set of isoforms. • Visually inspect the resulNng alignments. Seing a parameter a lifle too liberal or conservaNve can have a huge effect on alignment. • Consider running the same fastq files through mulNple alignment pipelines specific to each applicaNon. – Gene expression -‐> BowNe to transcriptome – Exon discovery -‐> RUM or other hybrid mapper – Variant detecNon -‐> GSNAP, GATK, Samtools mpileup • If your species has not been sequenced, use a de novo assembly method. Can also use the genome of a related species as a scaffold.
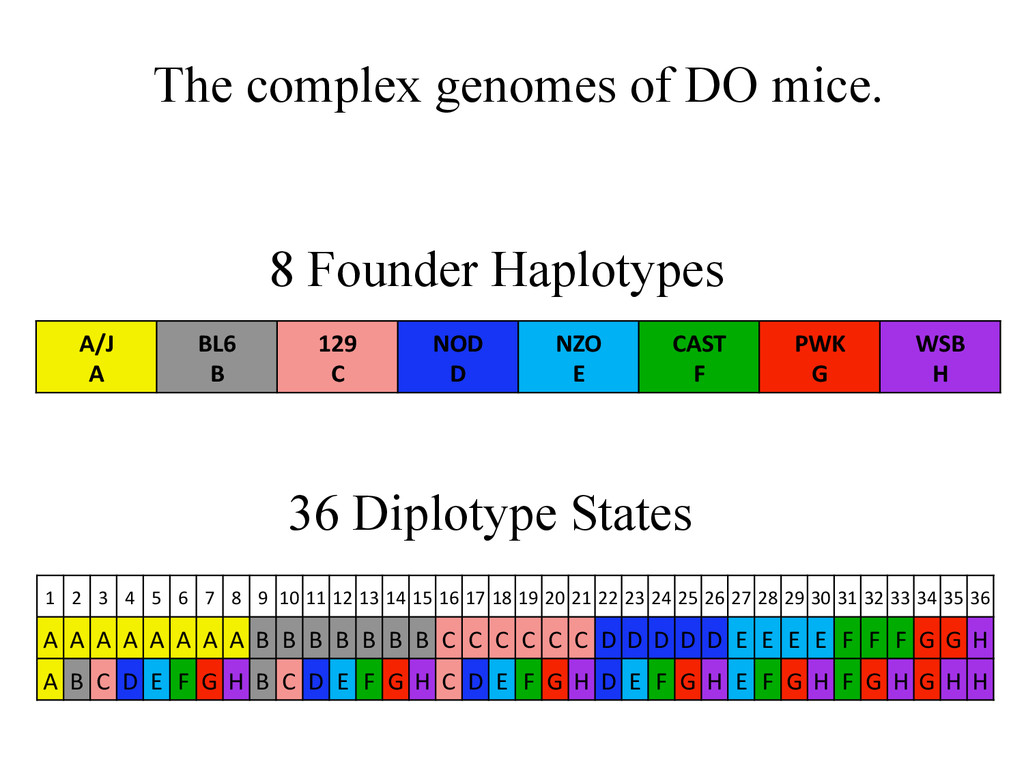
BL6 B 129 C NOD D NZO E CAST F PWK G WSB H 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 A A A A A A A A B B B B B B B C C C C C C D D D D D E E E E F F F G G H A B C D E F G H B C D E F G H C D E F G H D E F G H E F G H F G H G H H 36 Diplotype States 8 Founder Haplotypes
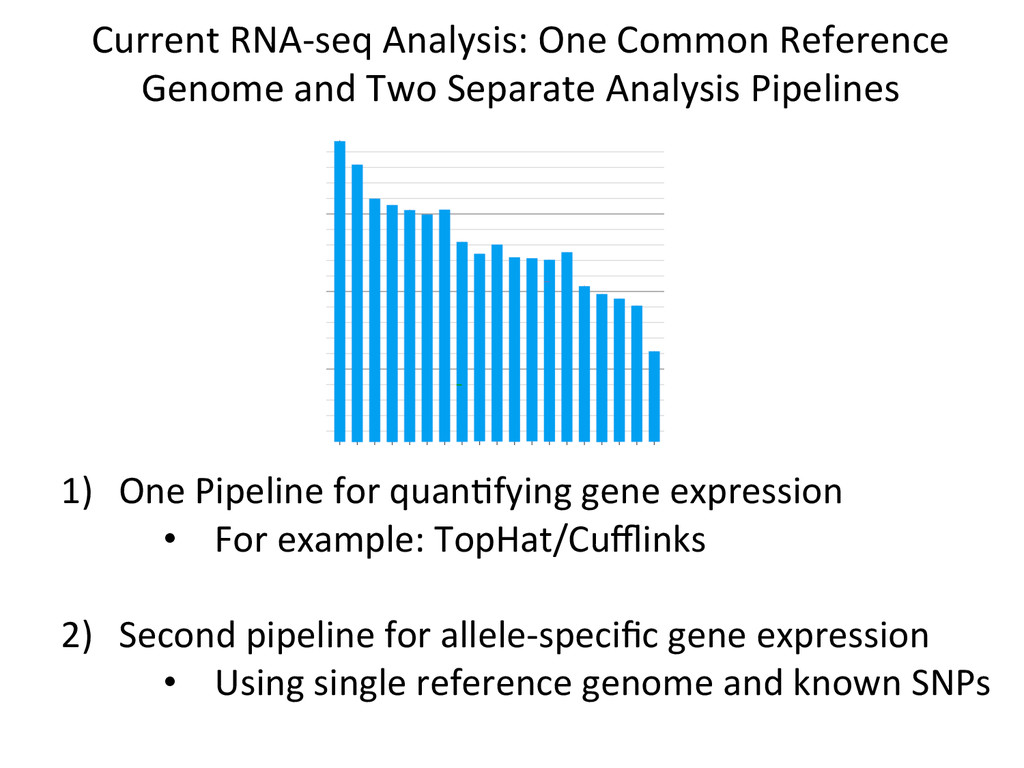
Separate Analysis Pipelines 1) One Pipeline for quanNfying gene expression • For example: TopHat/Cufflinks 2) Second pipeline for allele-‐specific gene expression • Using single reference genome and known SNPs
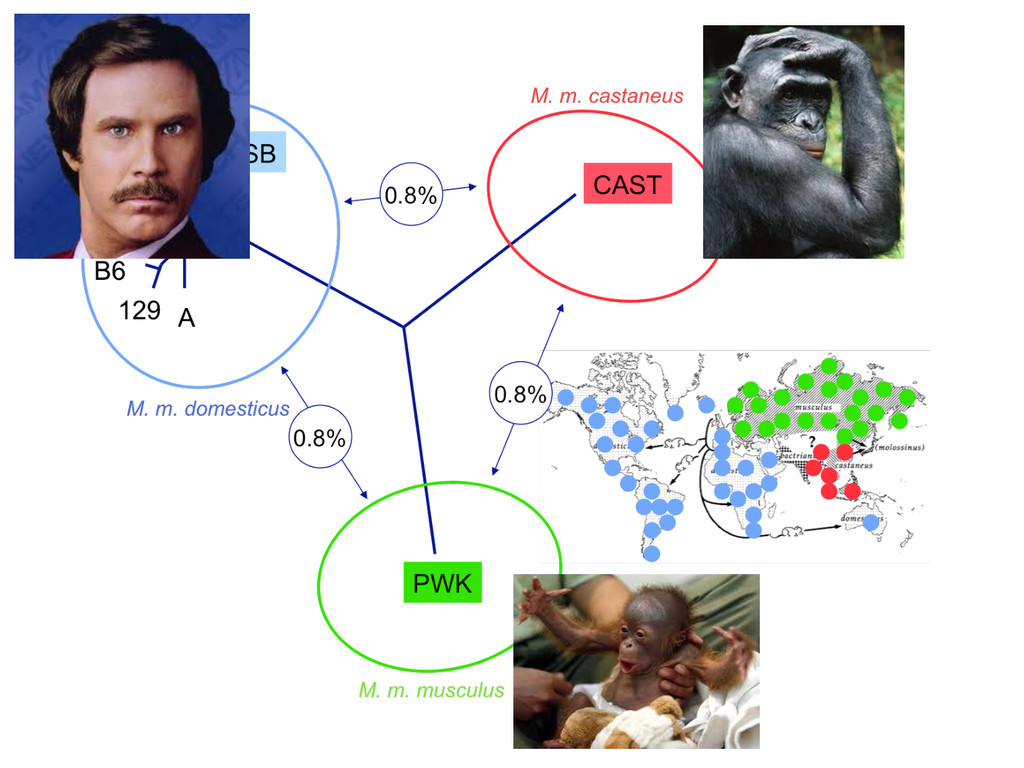
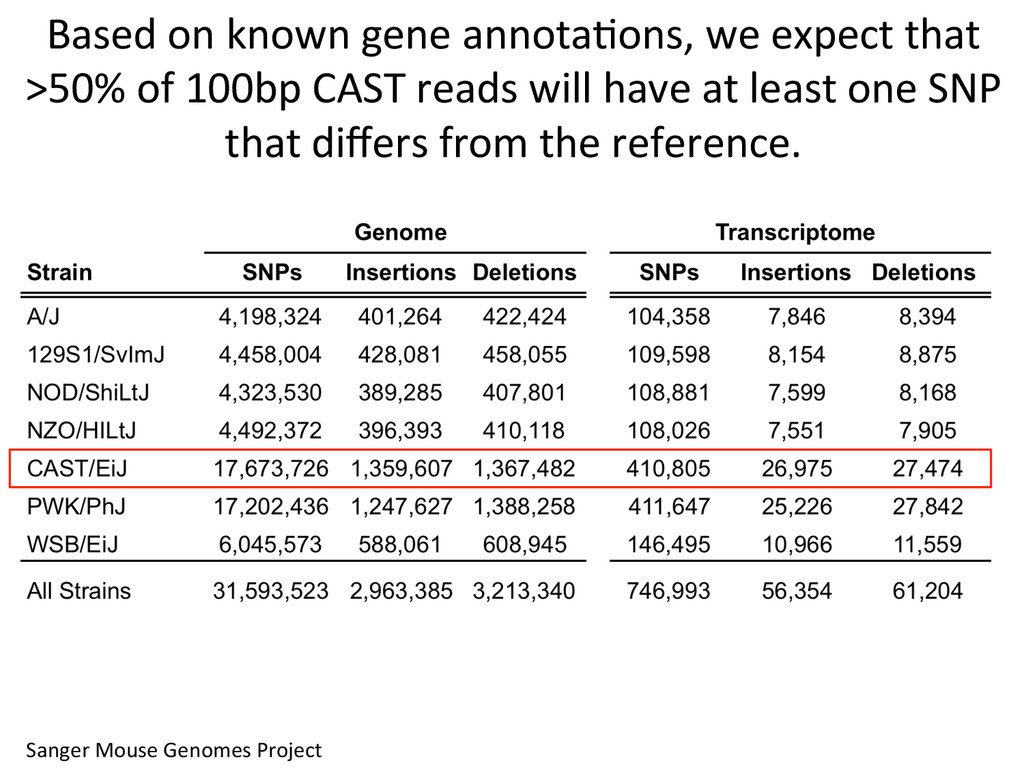
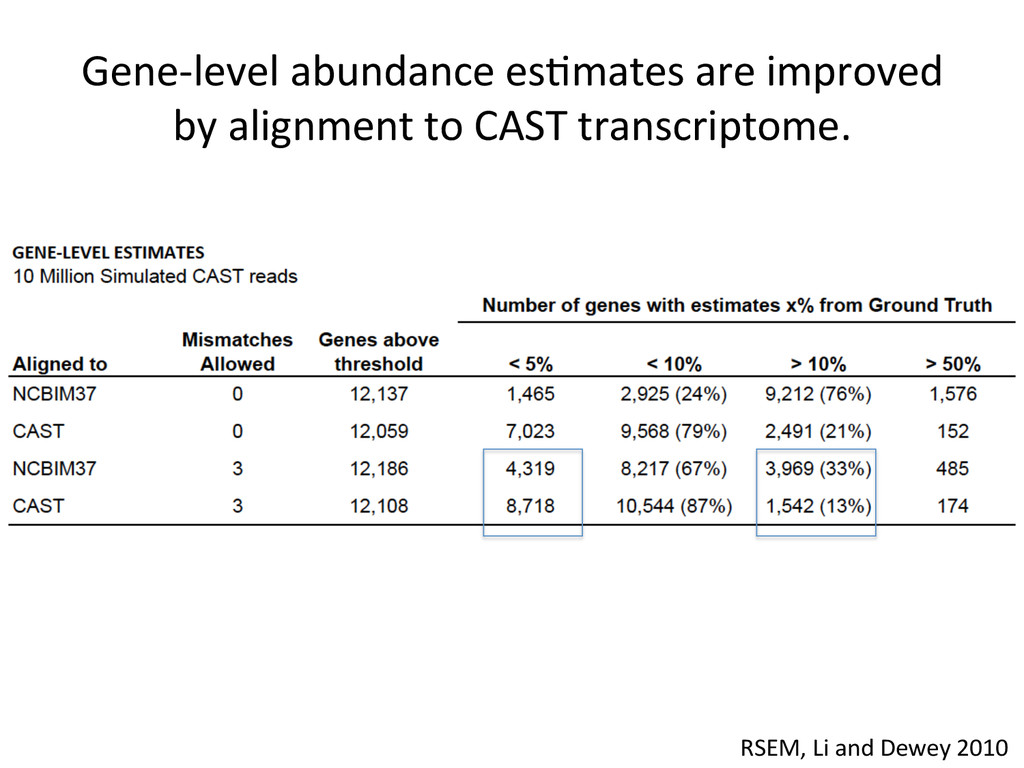
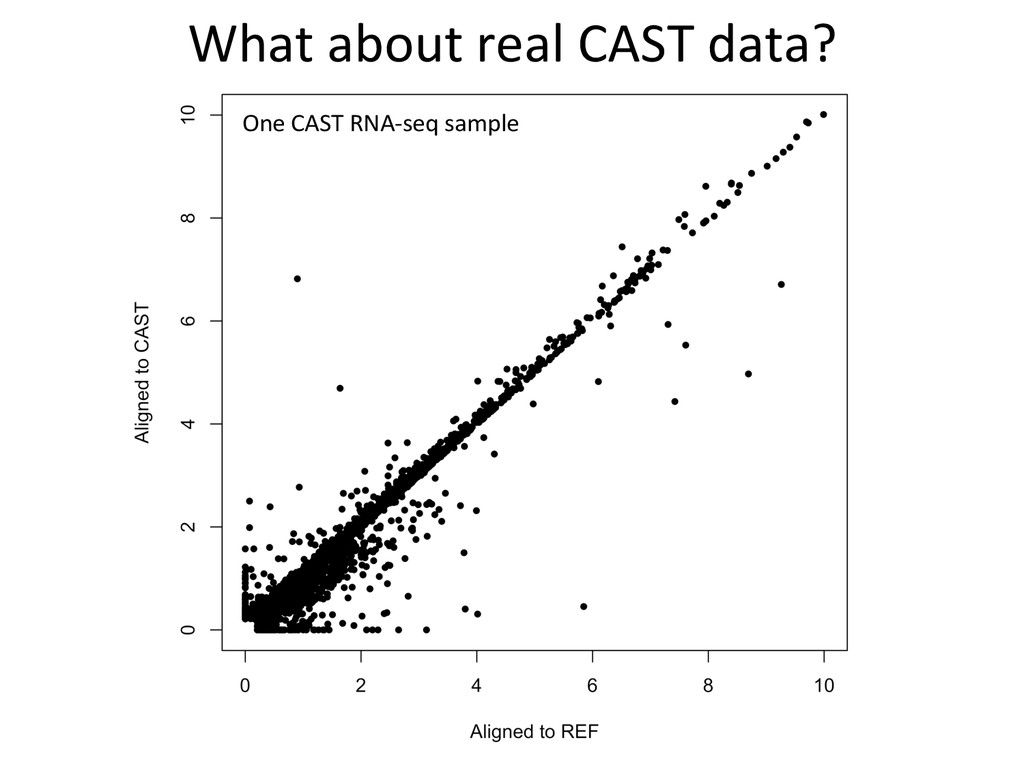
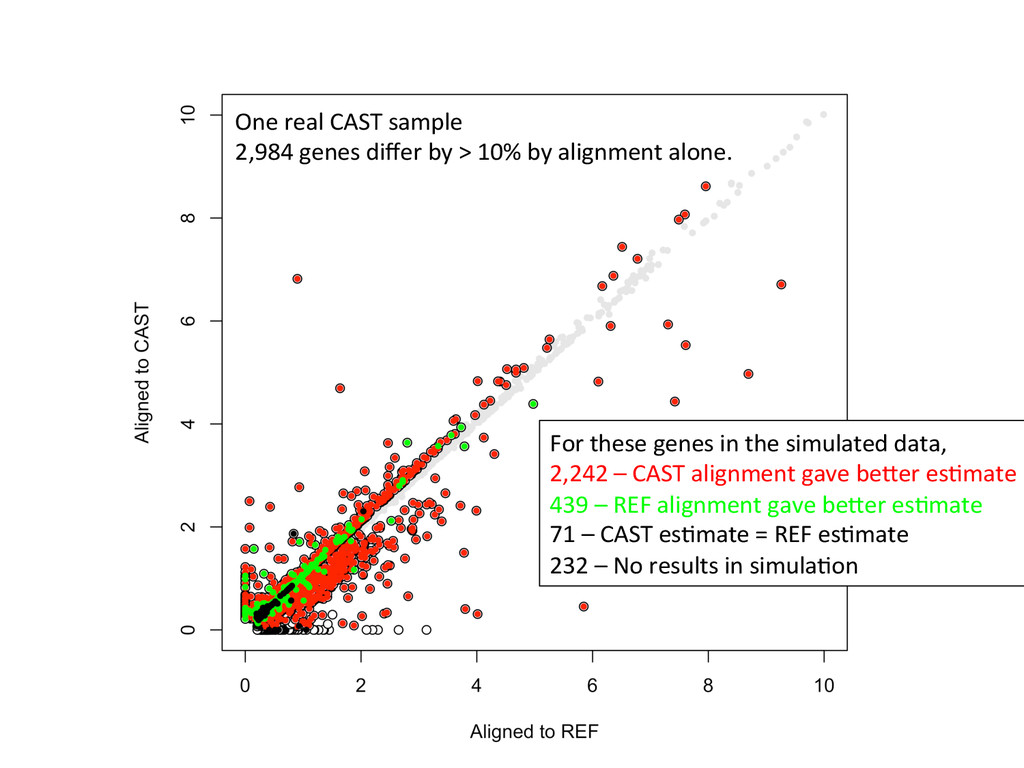
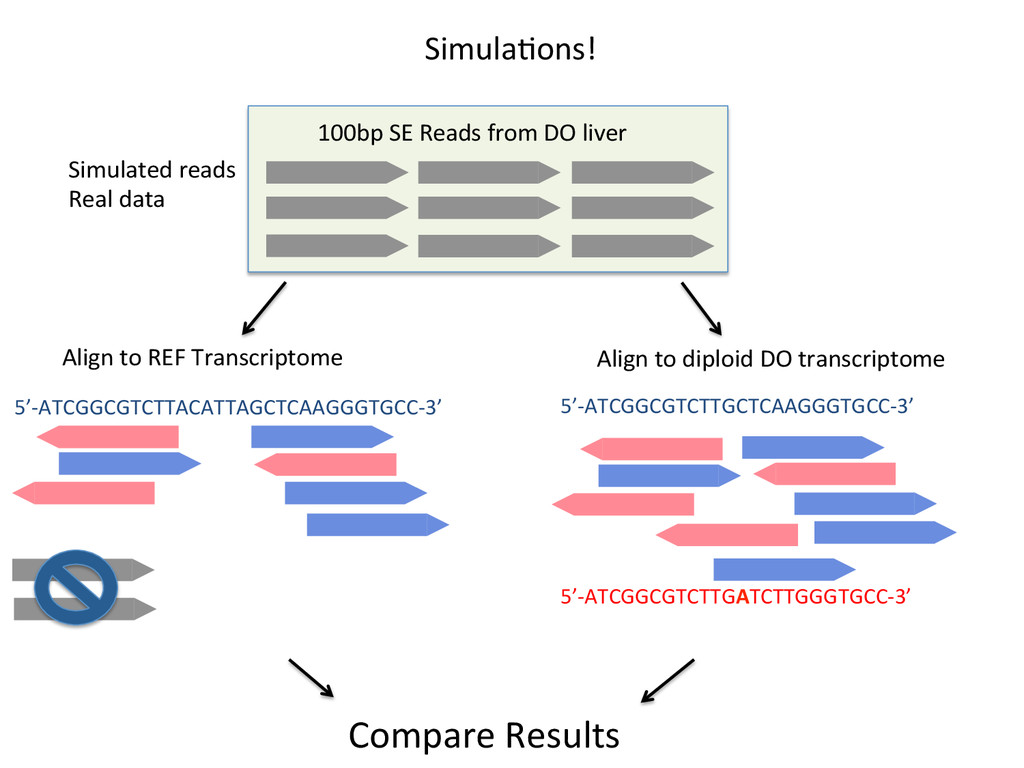
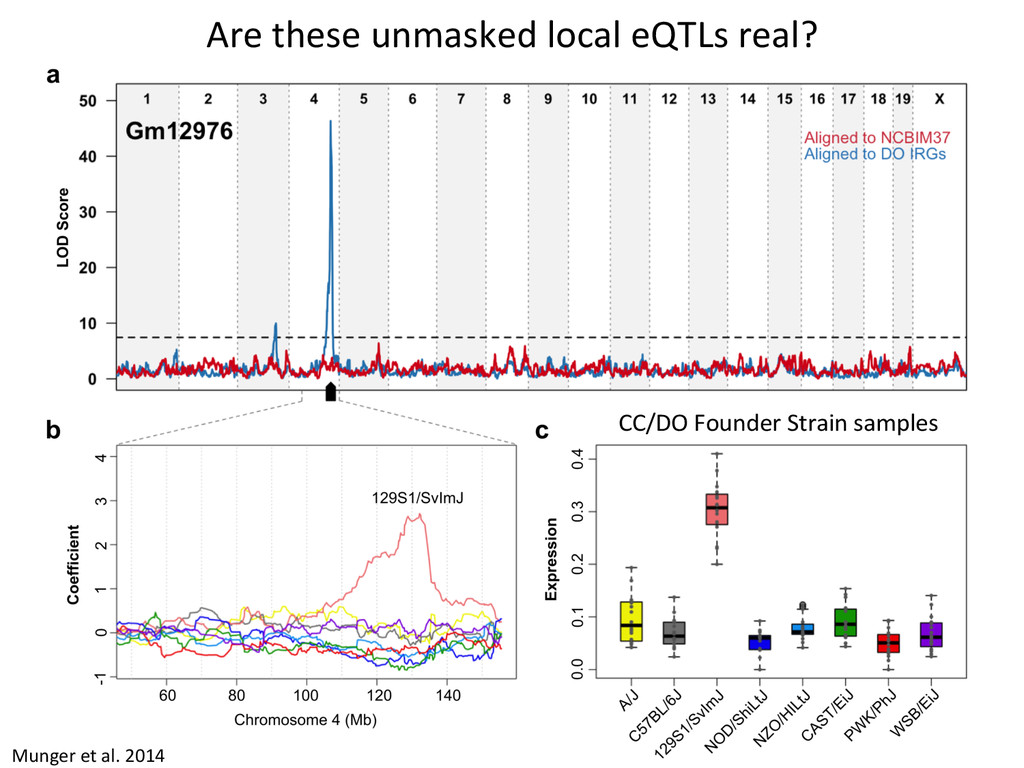
to ground truth Align to CAST Pseudotranscriptome 5’-‐ATCGGCGTCTTACATTAGCTCAAGGGTGCC-‐3’ 5’-‐ATCGGCGTCTTGCTCAAGGGTGCC-‐3’ Align to B6 Transcriptome 5’-‐ATCGGCGTCTTACATTAGCTCAAGGGTGCC-‐3’ To what degree do these differences affect alignment of RNA-‐Seq reads and gene abundance esNmates? Simulated reads Real data
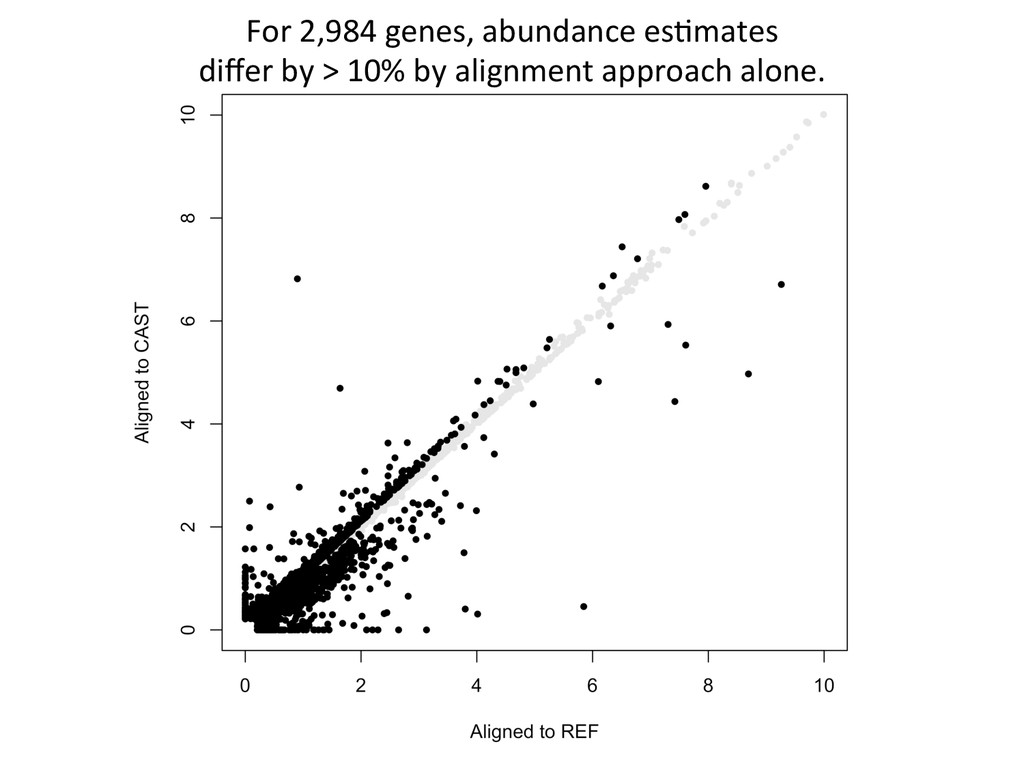
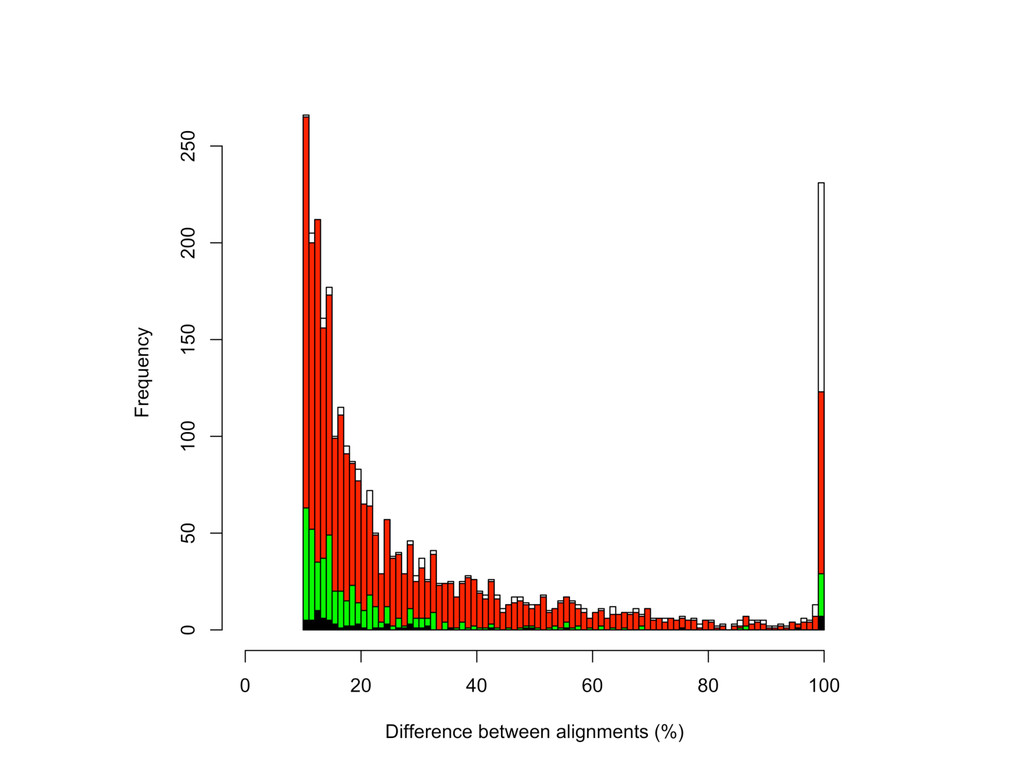
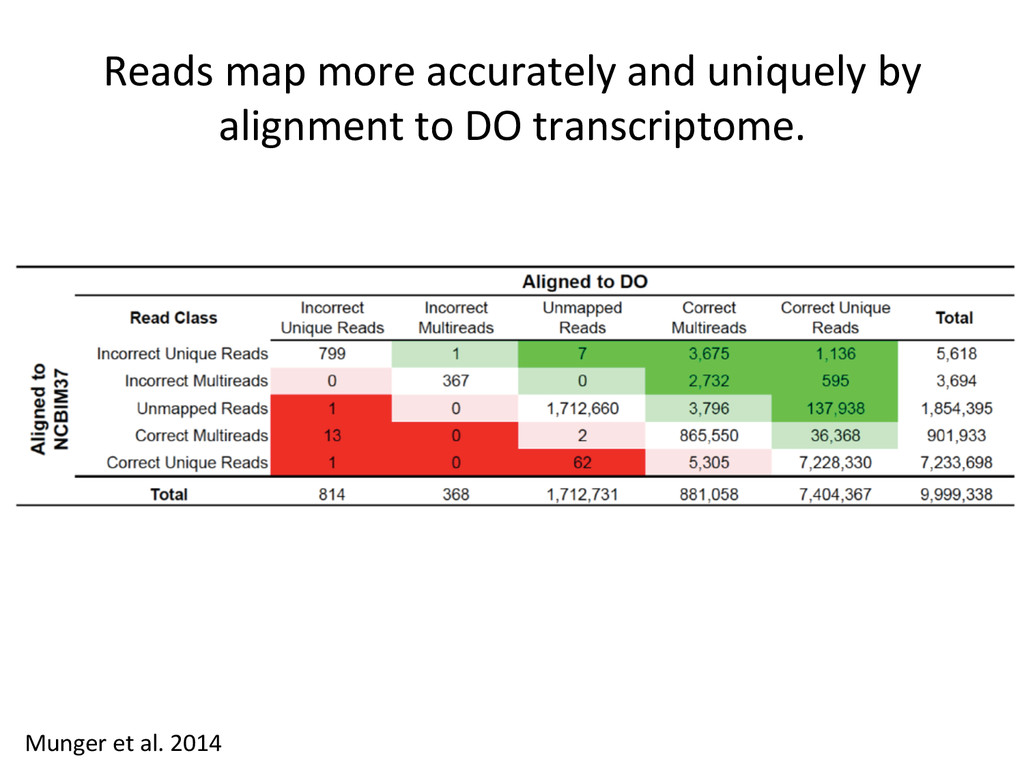
CAST alignment gave befer esNmate 439 – REF alignment gave befer esNmate 71 – CAST esNmate = REF esNmate 232 – No results in simulaNon One real CAST sample 2,984 genes differ by > 10% by alignment alone.
8 homozygous genotype states at every locus. A/J A BL6 B 129 C NOD D NZO E CAST F PWK G WSB H 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 A A A A A A A A B B B B B B B C C C C C C D D D D D E E E E F F F G G H A B C D E F G H B C D E F G H C D E F G H D E F G H E F G H F G H G H H Founder strains – 8 possible genotypes Diversity Outcross – 36 Possible Genotype states
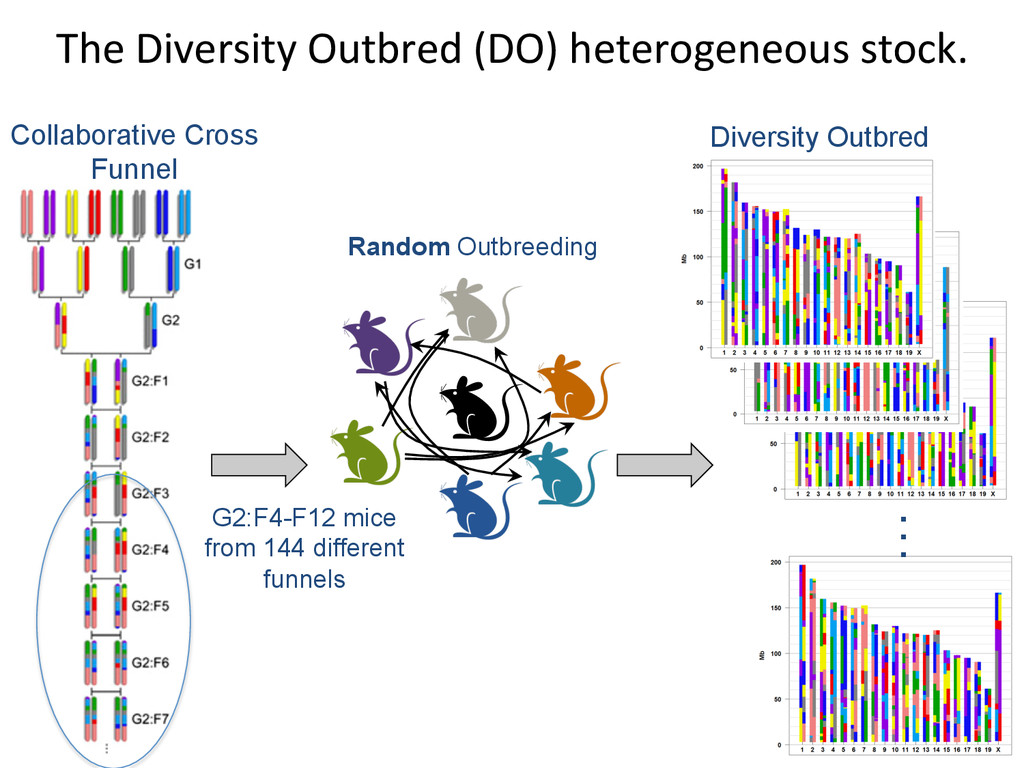
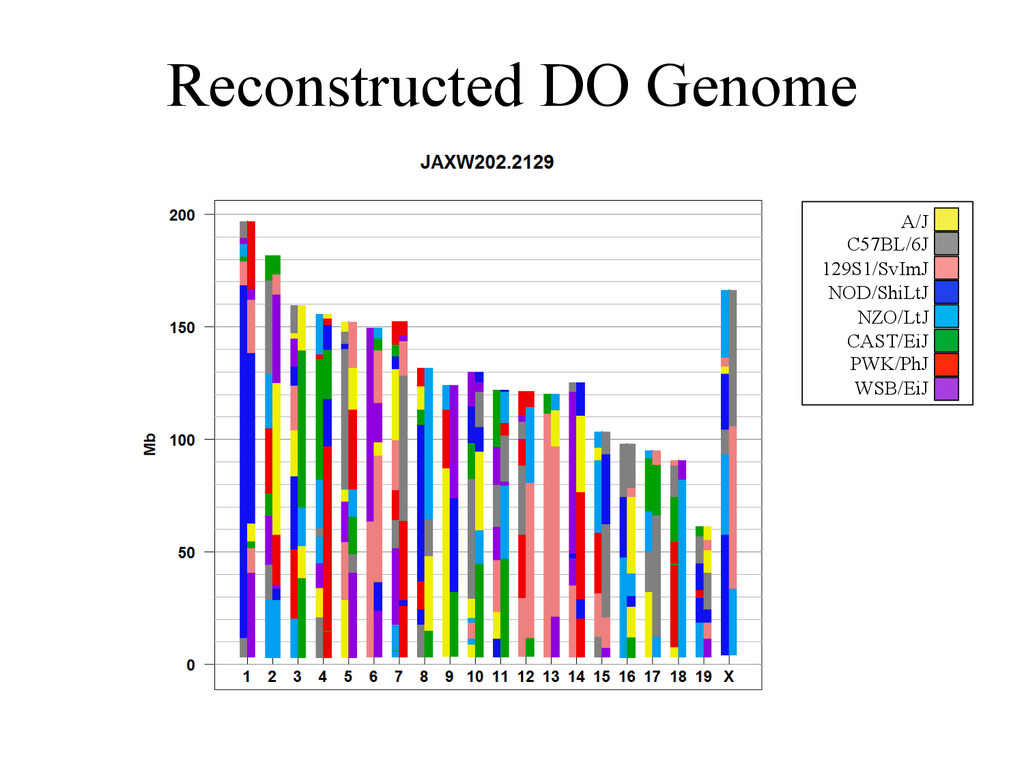
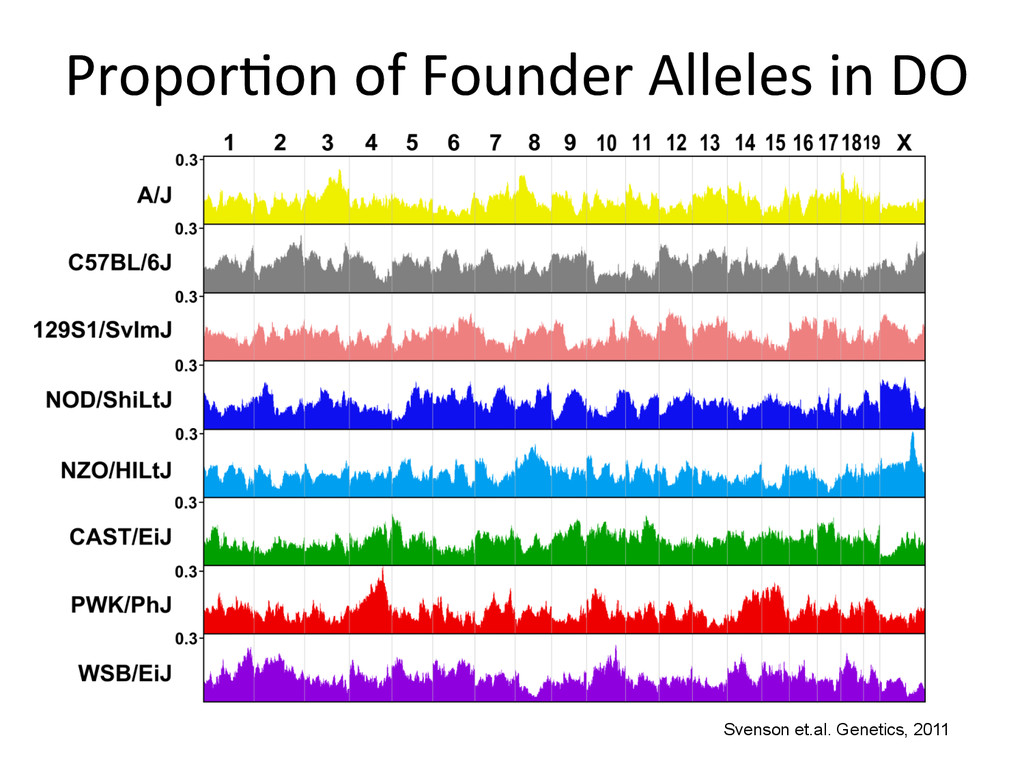
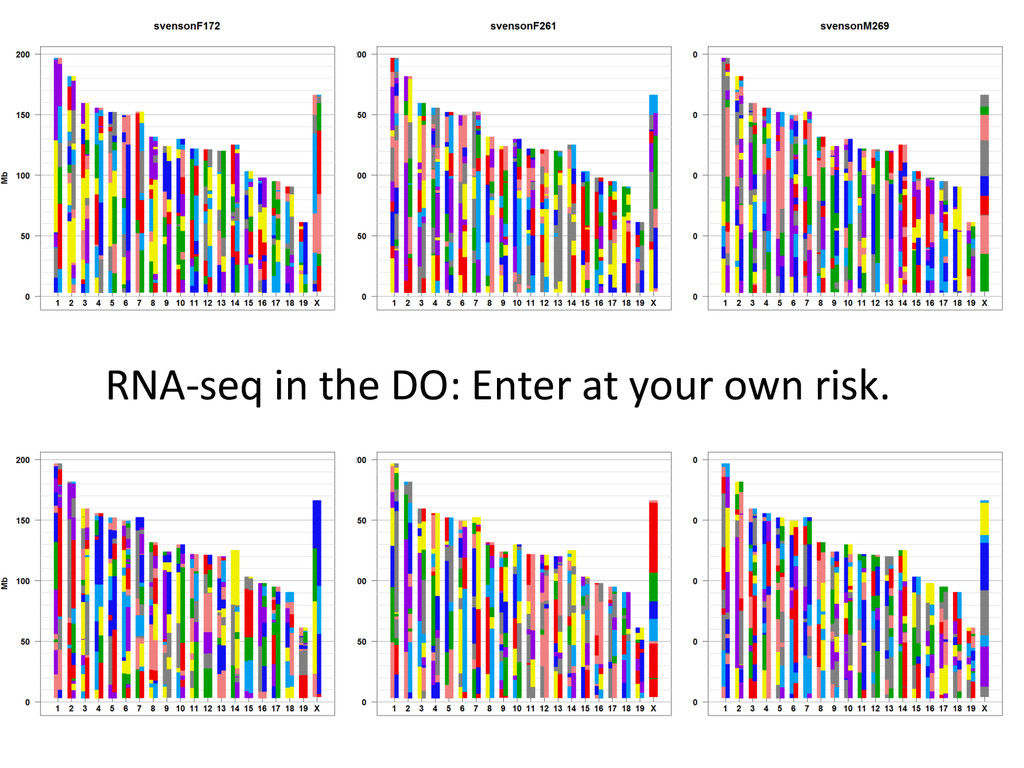
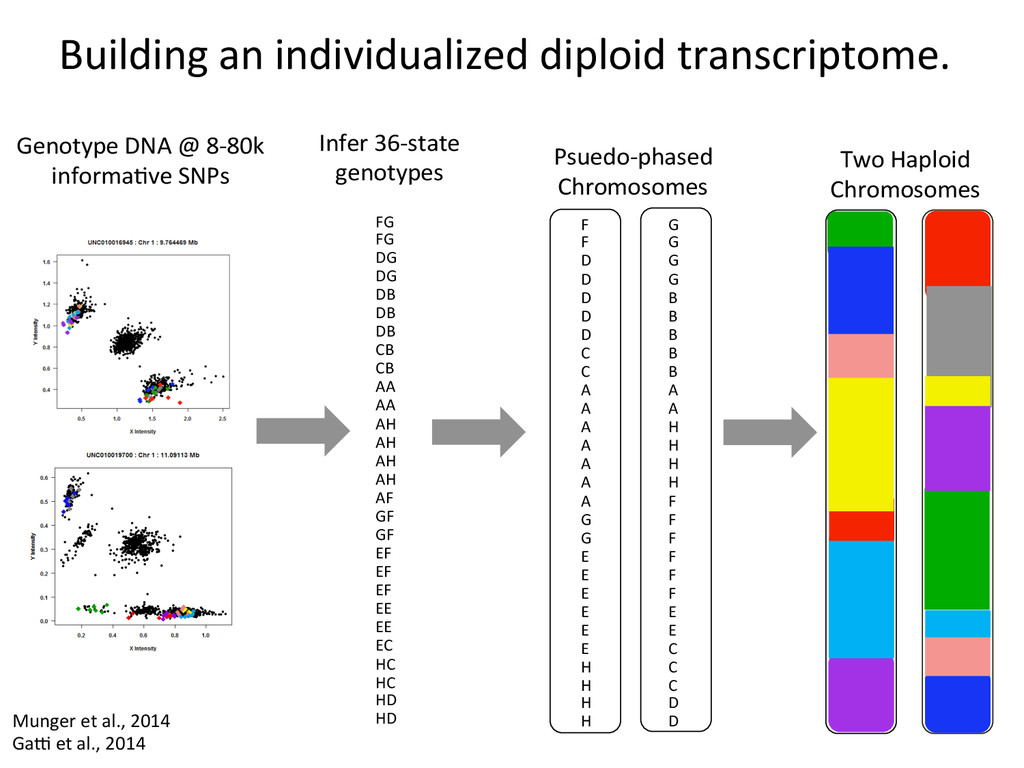
D D D D D C C A A A A A A A G G E E E E E E H H H H G G G G B B B B B A A H H H H F F F F F F E E C C C D D Psuedo-‐phased Chromosomes Two Haploid Chromosomes Genotype DNA @ 8-‐80k informaNve SNPs FG FG DG DG DB DB DB CB CB AA AA AH AH AH AH AF GF GF EF EF EF EE EE EC HC HC HD HD Infer 36-‐state genotypes Munger et al., 2014 Gai et al., 2014
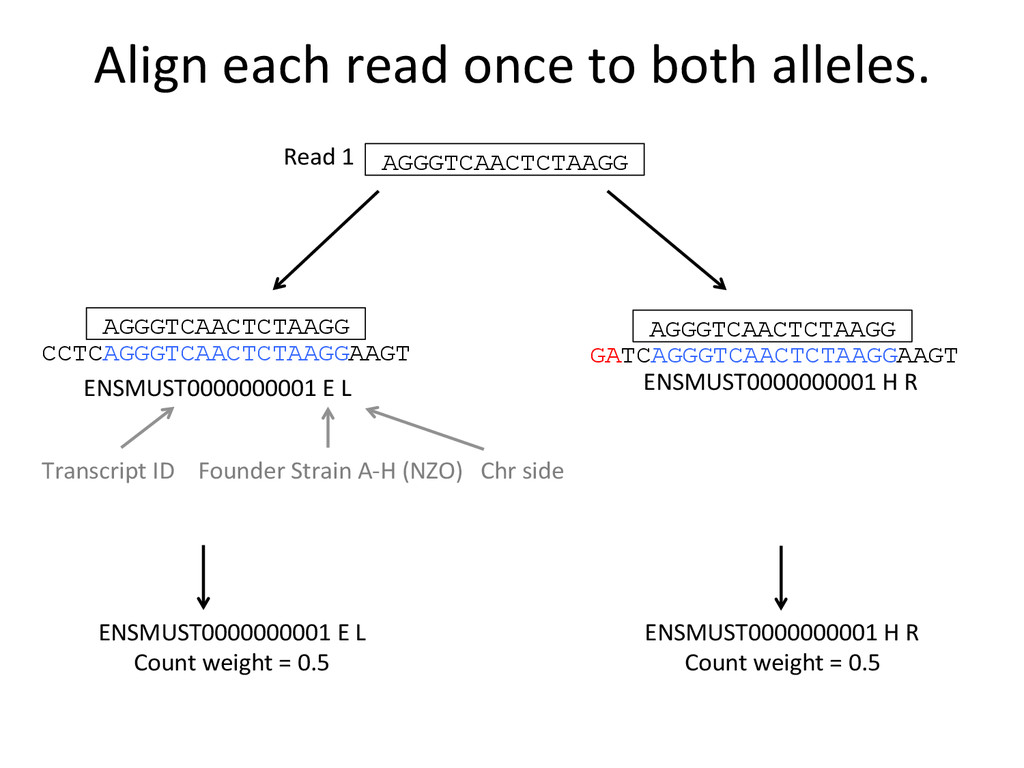
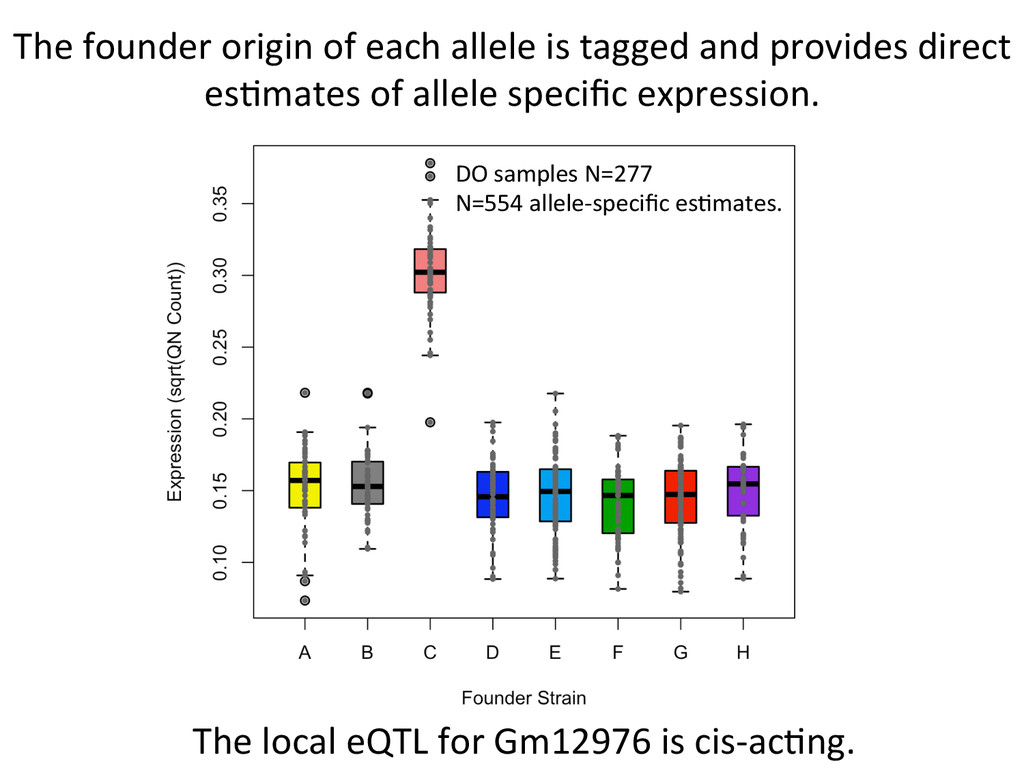
ENSMUST0000000001 E L ENSMUST0000000001 H R Transcript ID Founder Strain A-‐H (NZO) Chr side AGGGTCAACTCTAAGG Read 1 AGGGTCAACTCTAAGG AGGGTCAACTCTAAGG ENSMUST0000000001 E L Count weight = 0.5 ENSMUST0000000001 H R Count weight = 0.5
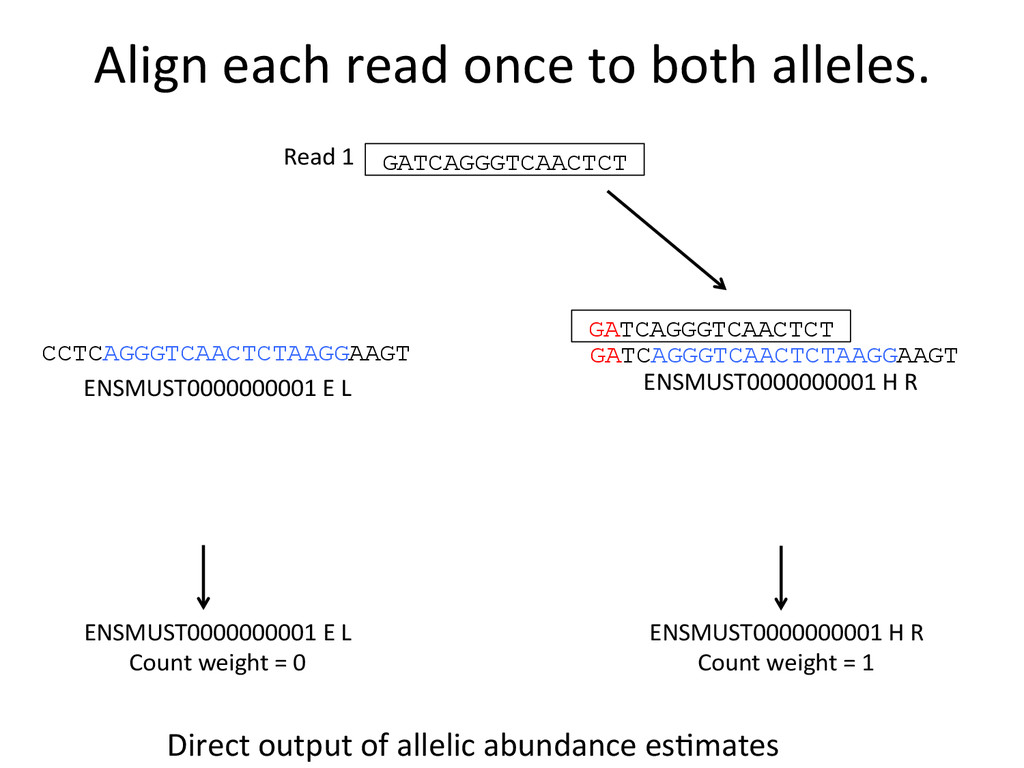
ENSMUST0000000001 E L ENSMUST0000000001 H R GATCAGGGTCAACTCT Read 1 GATCAGGGTCAACTCT ENSMUST0000000001 E L Count weight = 0 ENSMUST0000000001 H R Count weight = 1 Direct output of allelic abundance esNmates
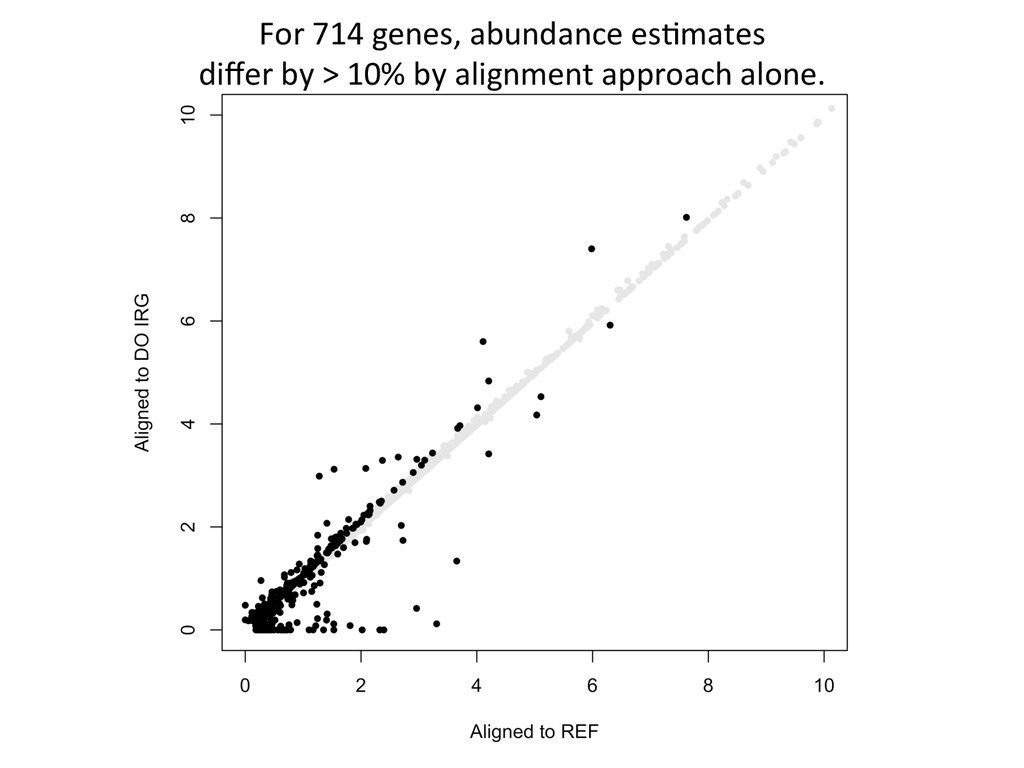
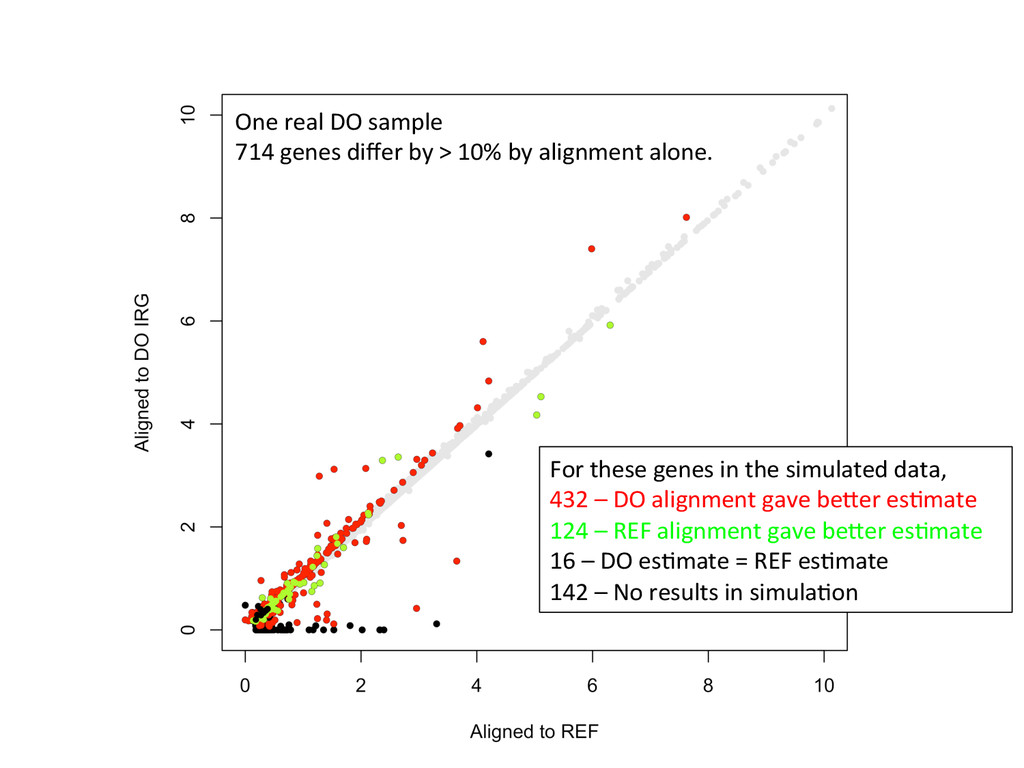
Align to diploid DO transcriptome 5’-‐ATCGGCGTCTTGCTCAAGGGTGCC-‐3’ 5’-‐ATCGGCGTCTTGATCTTGGGTGCC-‐3’ Align to REF Transcriptome 5’-‐ATCGGCGTCTTACATTAGCTCAAGGGTGCC-‐3’ SimulaNons! Simulated reads Real data
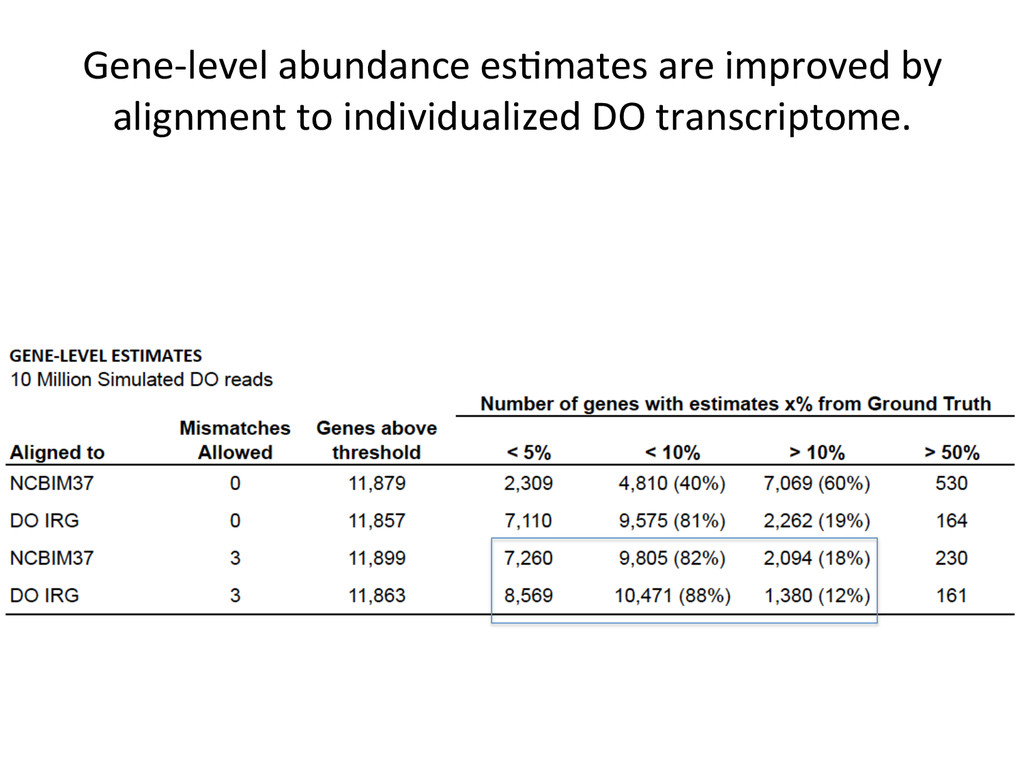
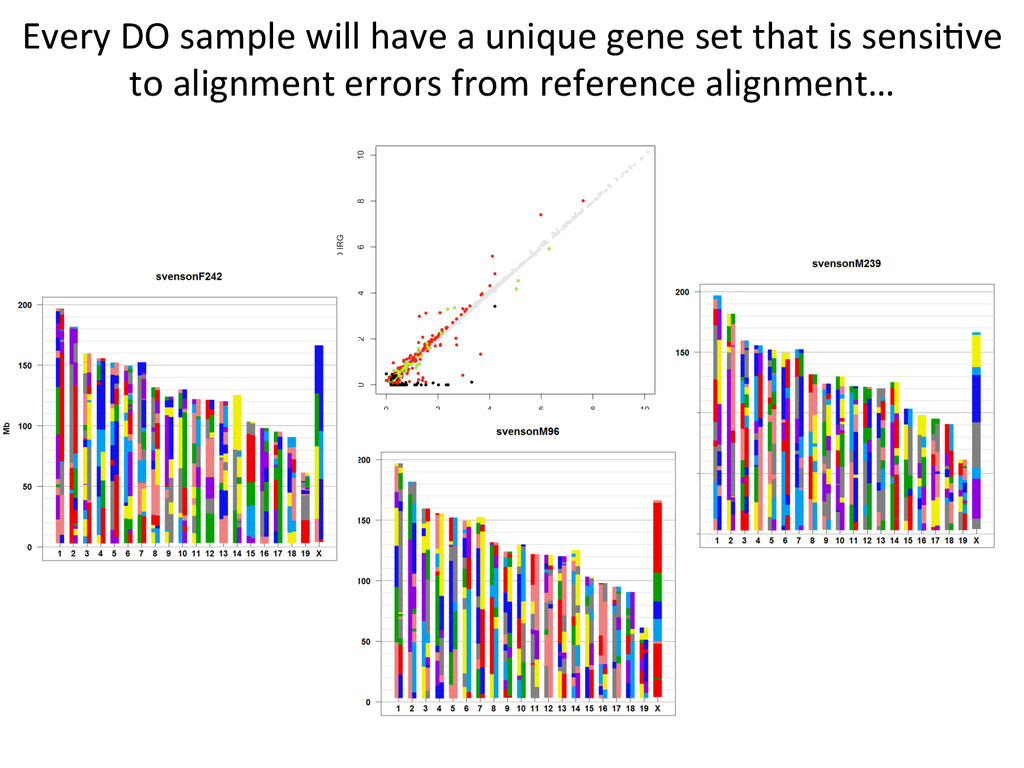
10% by alignment alone. For these genes in the simulated data, 432 – DO alignment gave befer esNmate 124 – REF alignment gave befer esNmate 16 – DO esNmate = REF esNmate 142 – No results in simulaNon
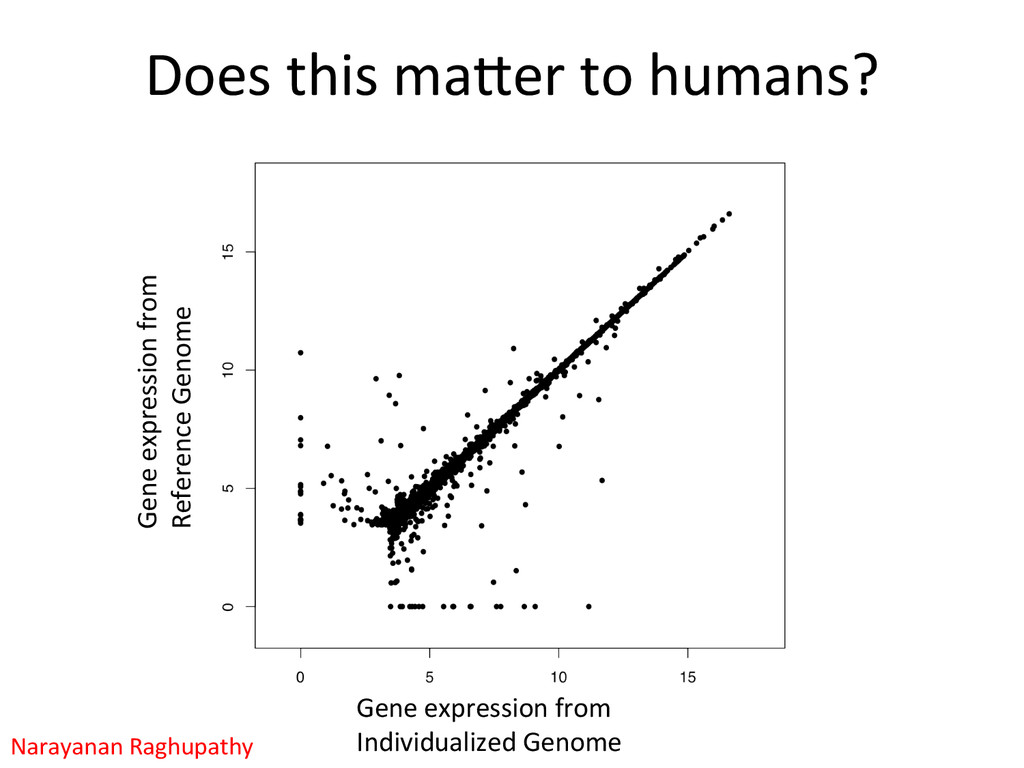
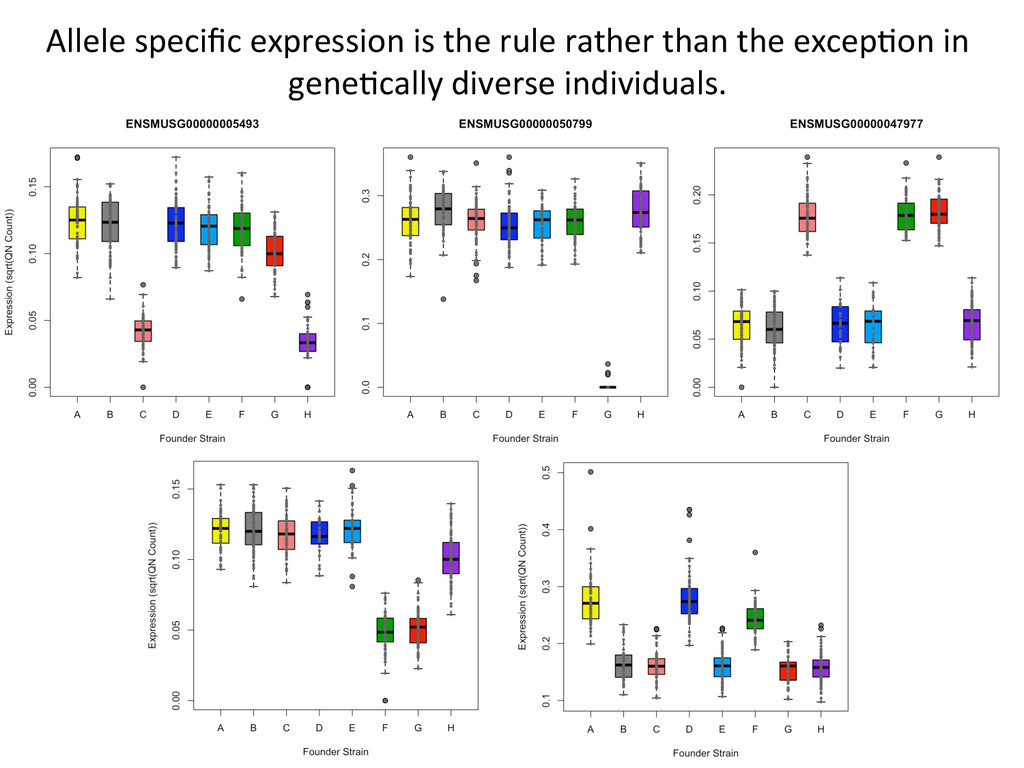
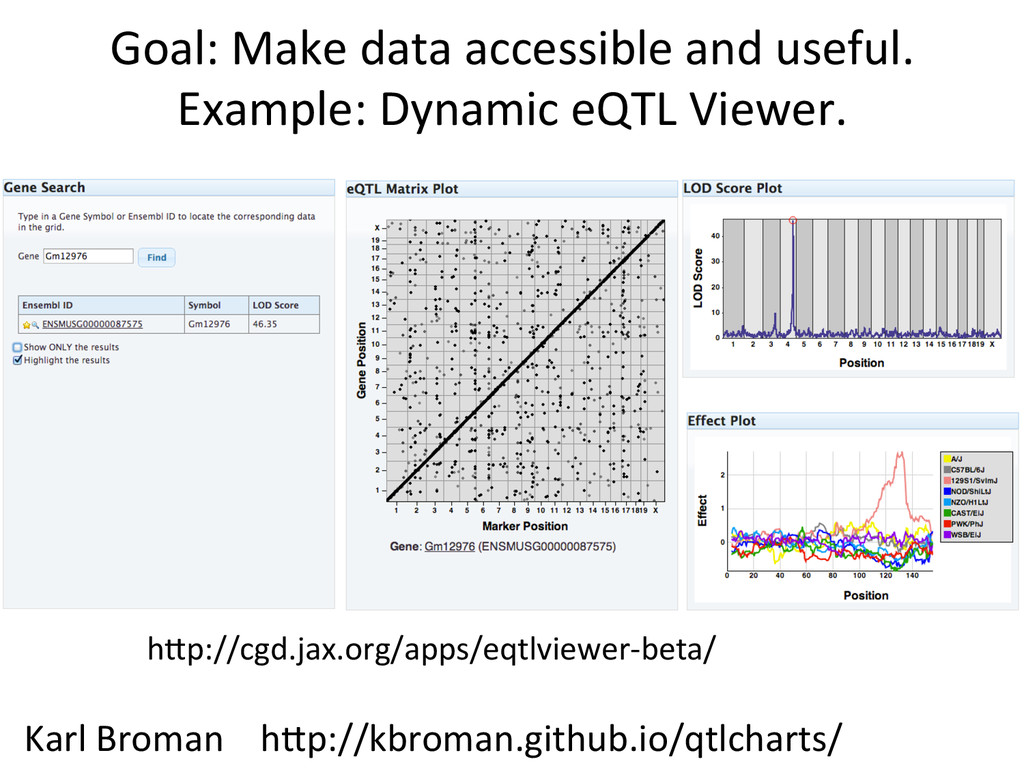
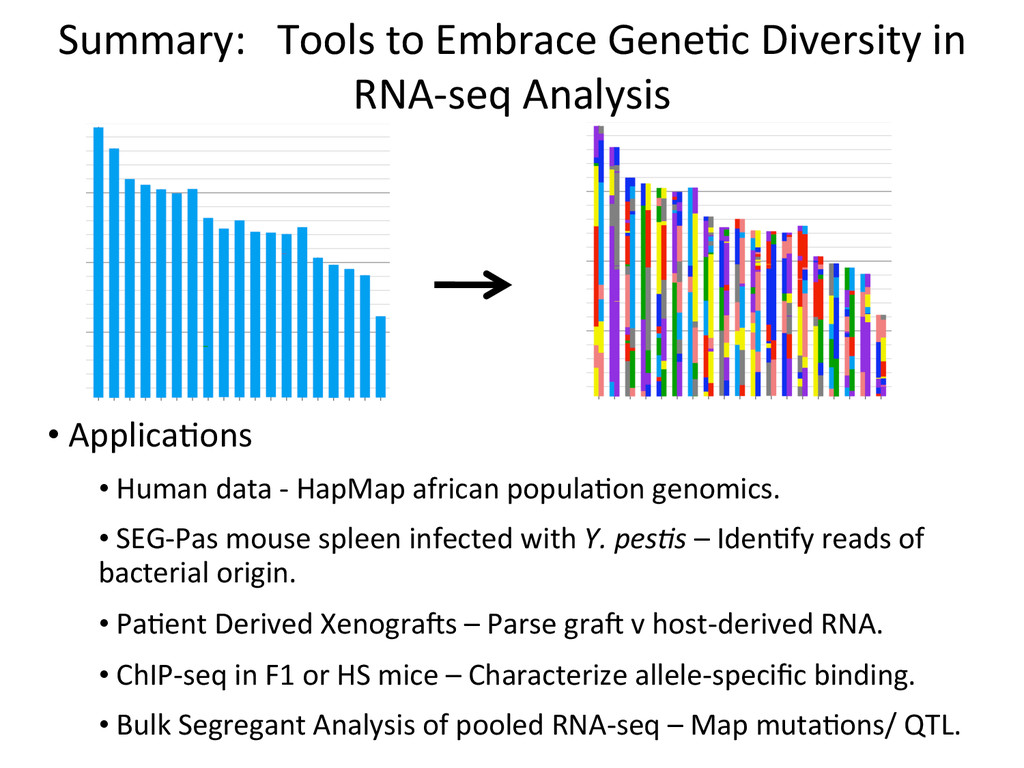
• Seqnature: Building personal diploid genomes using known geneNc variaNon. Can be applied to human data. • EMASE: Accurate and simultaneous quanNtaNon of gene expression and allele-‐specific expression • Diversity Outbred mice are ideal for high-‐resoluNon mapping of expression QTL. r/DOQTL: comprehensive DO analysis tools. • We have developed methodology for dealing with exploiGng the high geneNc diversity in this populaNon.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}