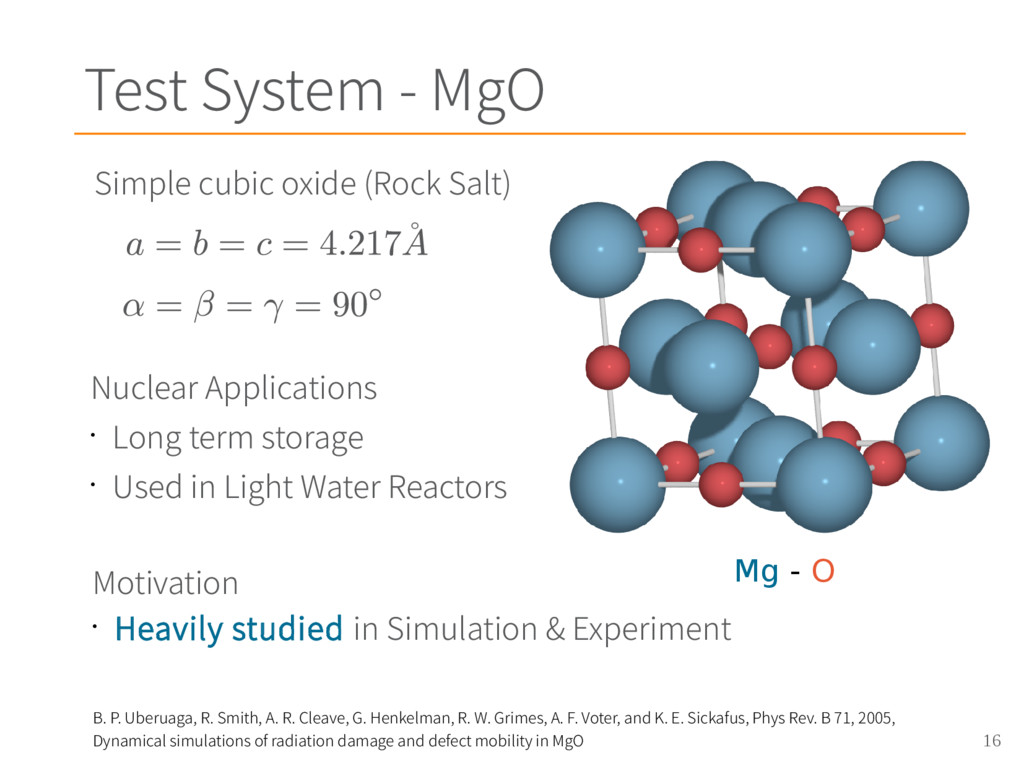
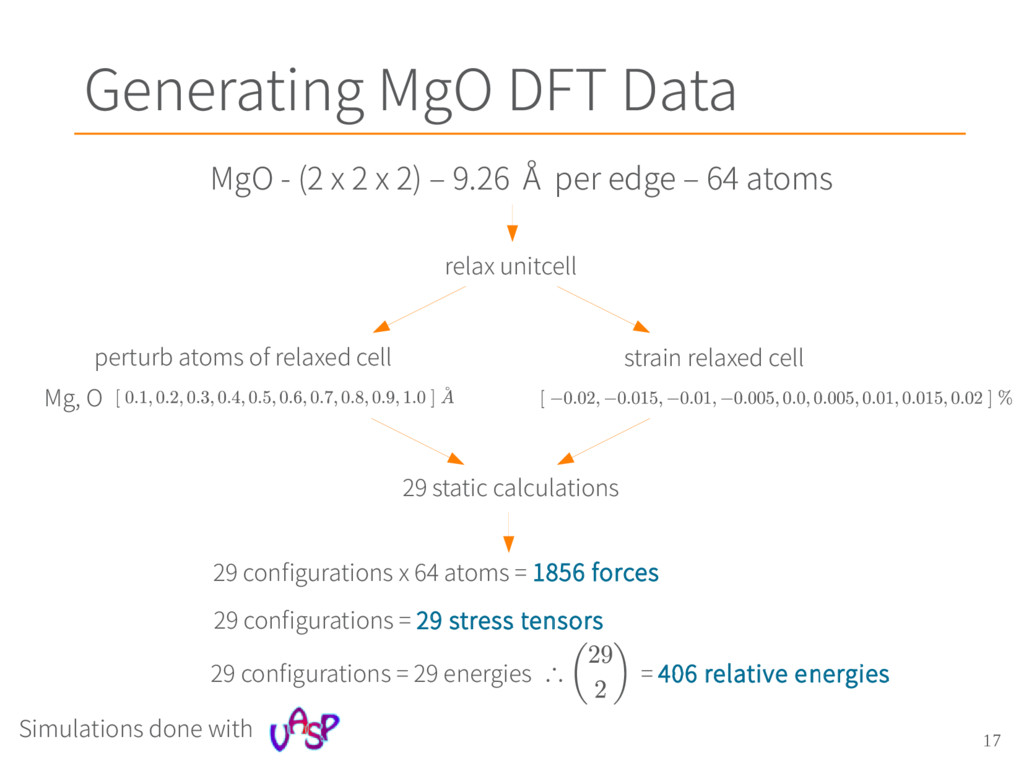
Smith, A. R. Cleave, G. Henkelman, R. W. Grimes, A. F. Voter, and K. E. Sickafus, Phys Rev. B 71, 2005, Dynamical simulations of radiation damage and defect mobility in MgO [2] – Masanori Matsui, J. Chem. Phys. 91, 489 (1989), Molecular dynamics study of the structural and thermodynamic properties of MgO crystal with quantum correction [3] – G. V. Lewis and C. R. A. Catlow, J. Phys. C: Solid State Phys. 18 1149, (1985), Potential models for ionic oxides [4] – Graeme Henkelman, Blas P. Uberuaga, Duncan J. Harris, John H. Harding, and Neil L. Allan, Phys. Rev. B 72, 115437, 2005, MgO addimer diffusion on MgO(100): A comparison of ab initio and empirical models [5] - F. Ercolessi and J. B. Adams Europhys. Lett. 26 583, 1994 Interatomic Potentials from First-Principles Calculations: The Force-Matching Method [6] - Sergei Izvekov, Michele Parrinello, Christian J. Burnham and Gregory A. Voth, J. Chem. Phys. 120, 10896, 2004, Effective force fields for condensed phase systems from ab initio molecular dynamics simulation: A new method for force-matching [7] - Eric Jones, Travis Oliphant, Pearu Peterson and others., SciPy: Open source scientific tools for Python, www.scipy.org, 2001 MgO applications MgO potentials Force matching origin Beautiful force matching paper Least Square Solver Charge Density for LiNbO 3 calculated with Quantum Espresso Acknowledgements • UTK Compute Cluster Newton • NERSC Super Computer Hopper • NICS Super Computer Darter All images and figures created by Chris Ostrouchov
{kind=link}
{kind=link}
{kind=link}
{kind=link}
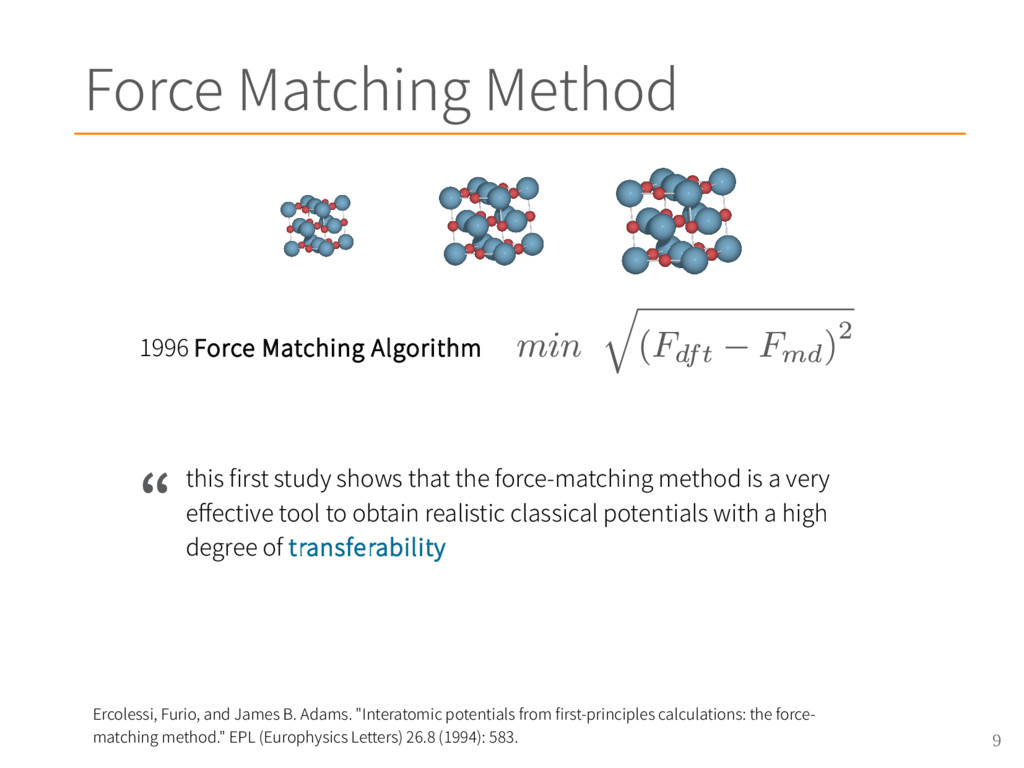
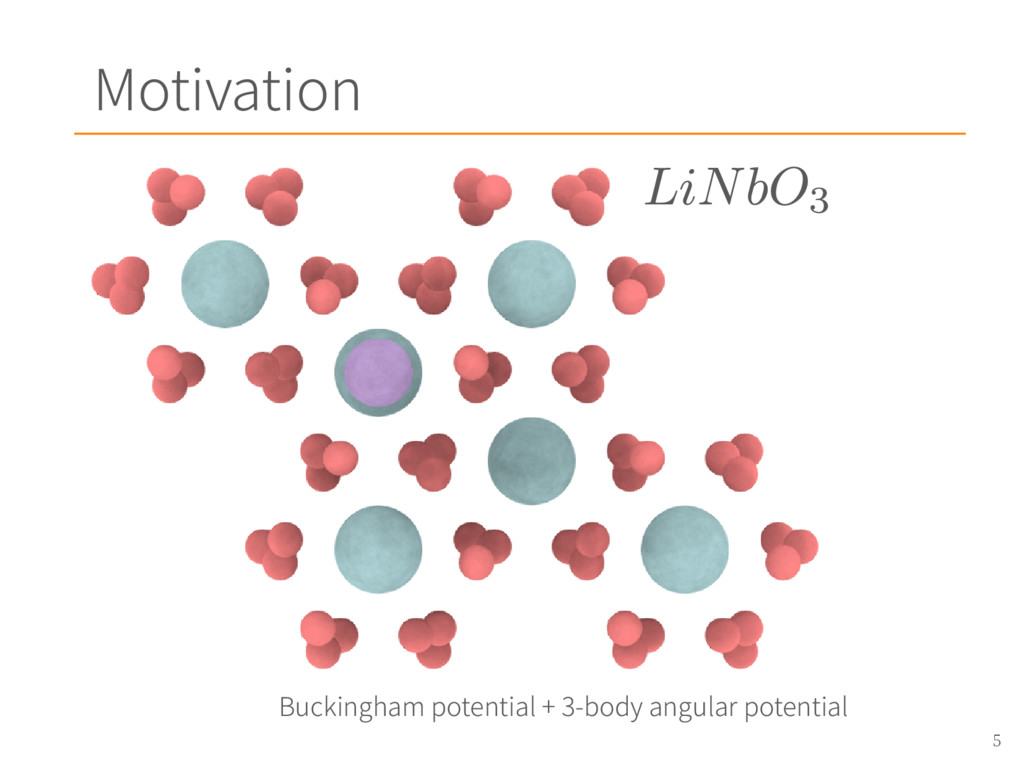{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![20 Potential Improvement a 0 [A] B 0 [GPa] E](https://files.speakerdeck.com/presentations/ddfe18cfc9c4472d9b17498236202b8b/slide_19.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![25 Thank You! References [1] – B. P. Uberuaga, R.](https://files.speakerdeck.com/presentations/ddfe18cfc9c4472d9b17498236202b8b/slide_24.jpg){kind=link}