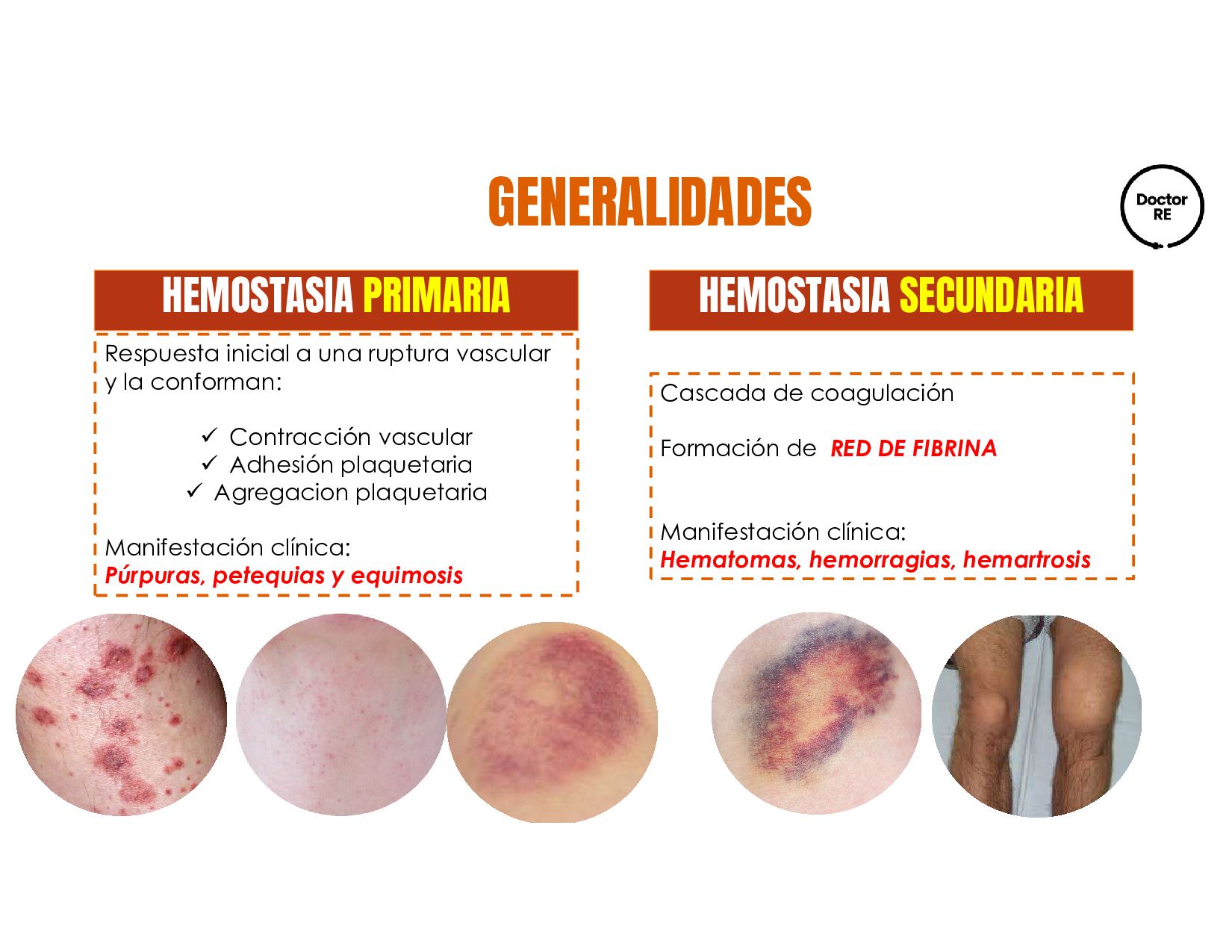
vascular y la conforman: ✓ Contracción vascular ✓ Adhesión plaquetaria ✓ Agregacion plaquetaria Manifestación clínica: Púrpuras, petequias y equimosis Cascada de coagulación Formación de RED DE FIBRINA Manifestación clínica: Hematomas, hemorragias, hemartrosis
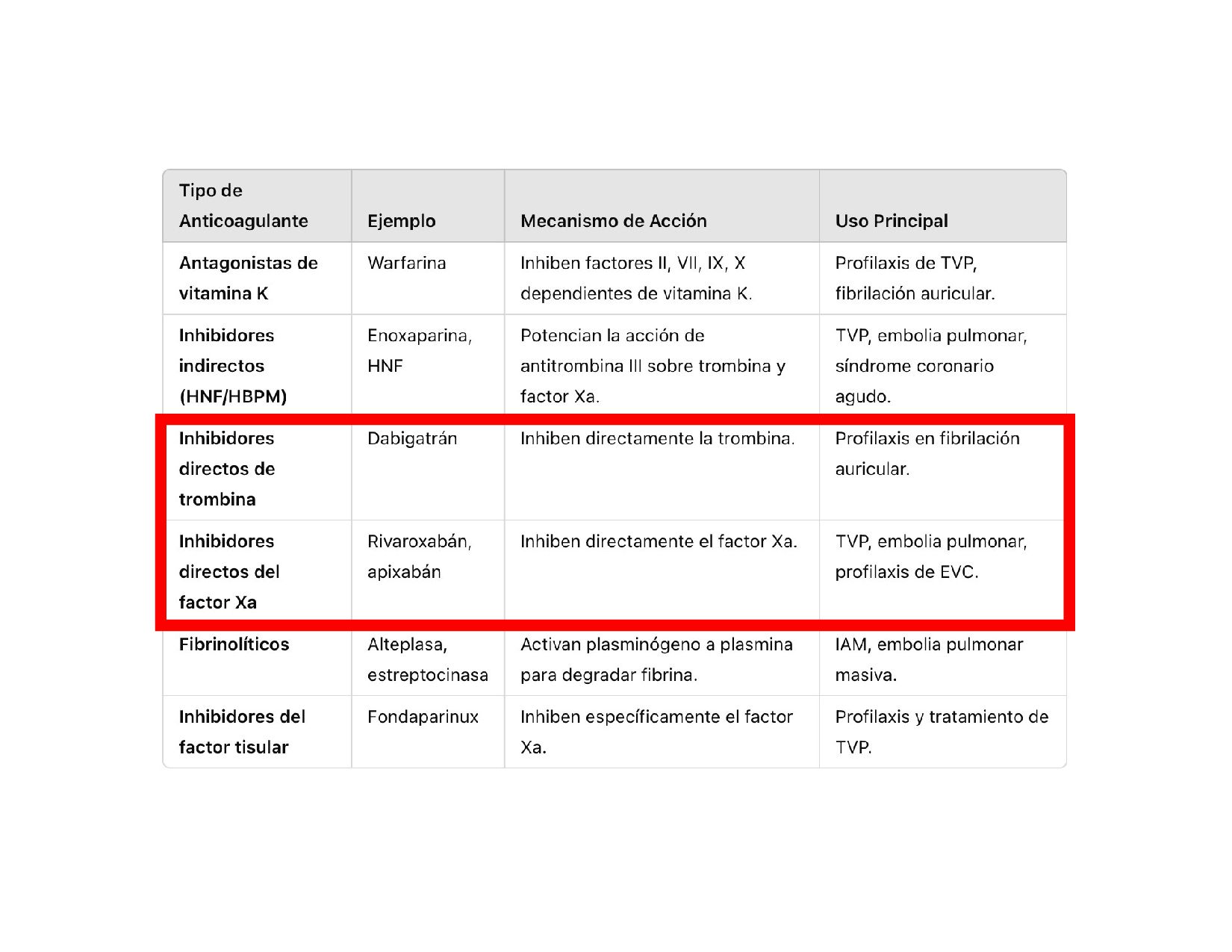
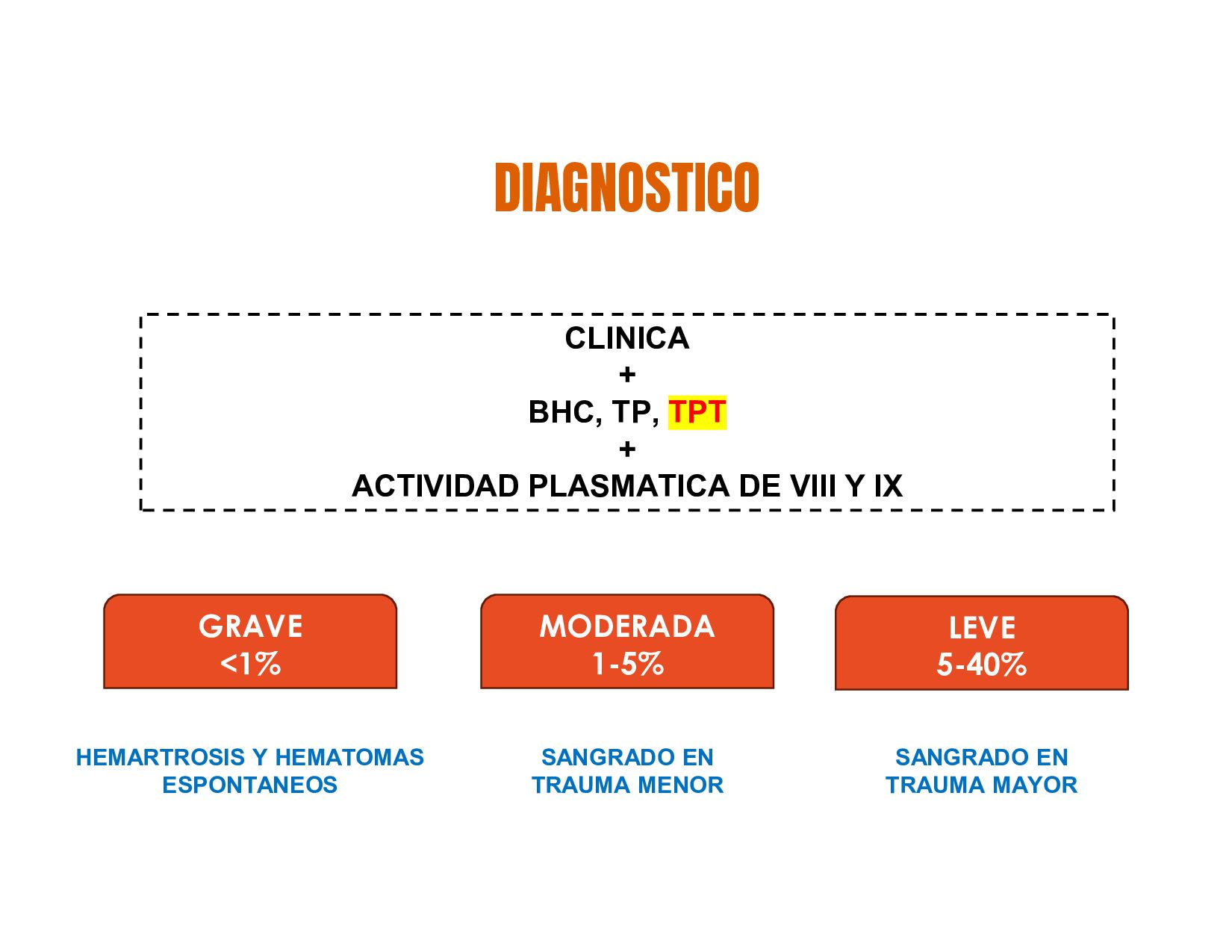
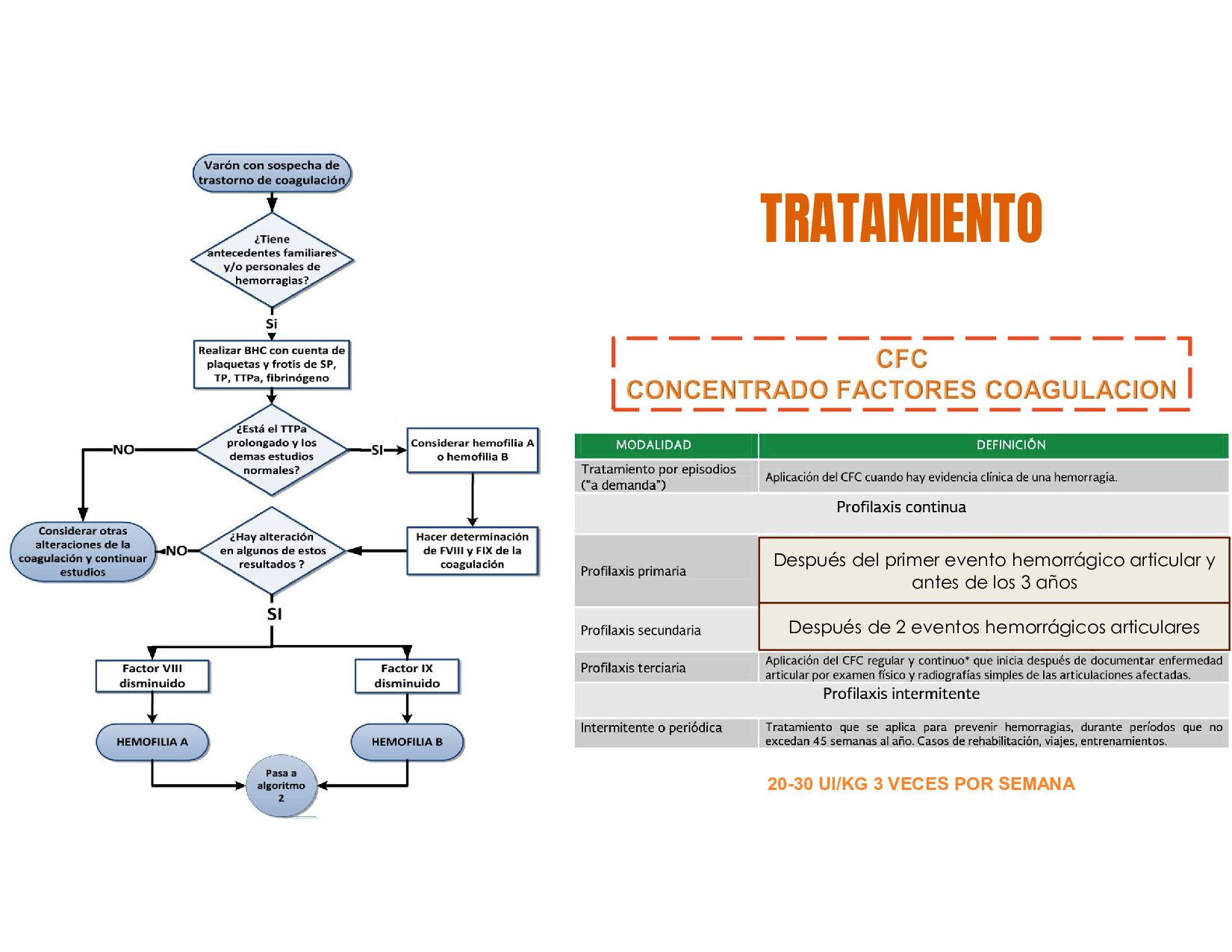
C, S ALTERACIÓN FACTOR VIII = HEMOFILIA Ab ALTERACIÓN FACTOR IX = HEMOFILIA B TP = EVALUA LA VIA EXTRINSECA, WARFARINA Y FACTORES VITAMINA K DEPENDIENTES TPT= VIA INTRINSECA, HEPARINA, COAGULOPATIAS HEREDITARIAS Proteólisis HEMOSTASIA SECUNDARIA
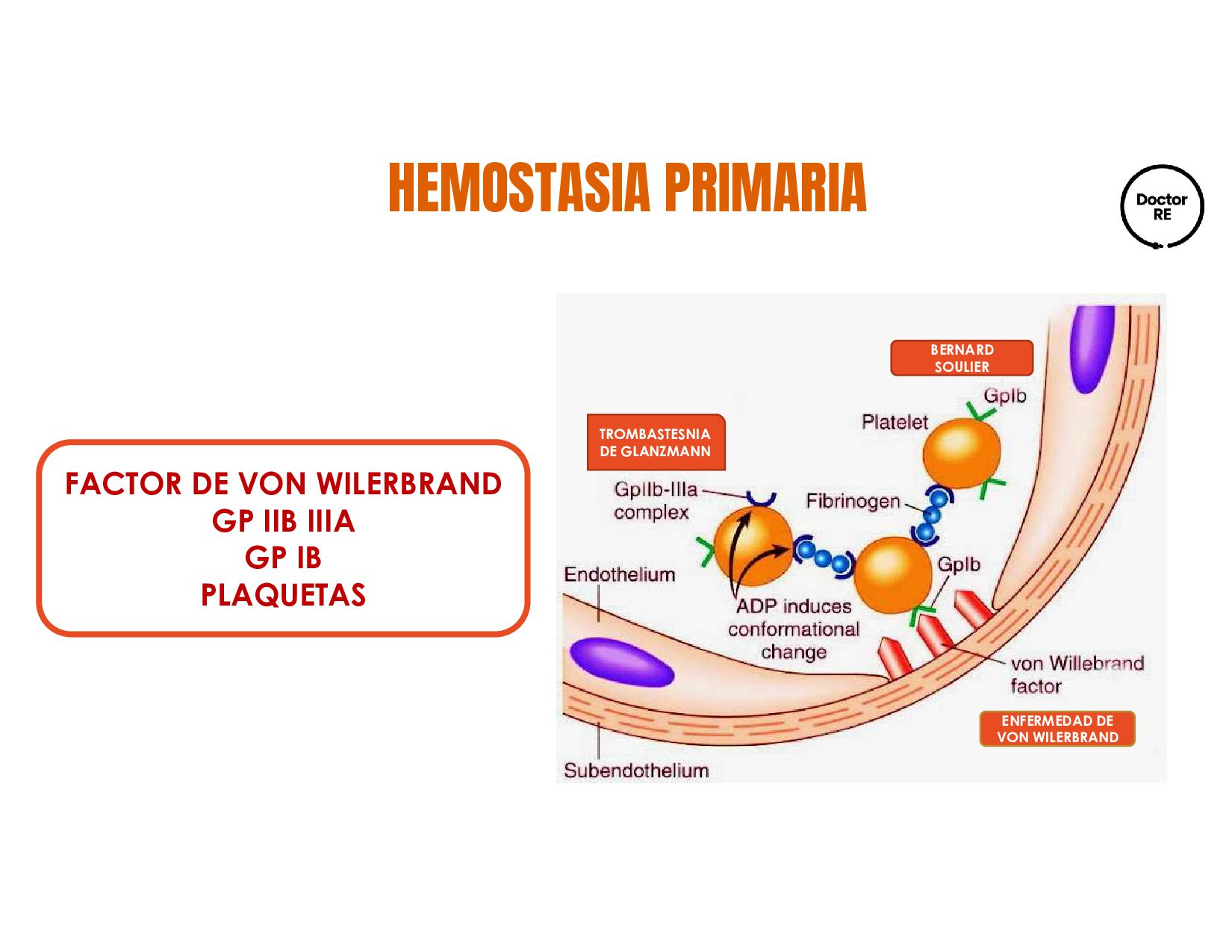
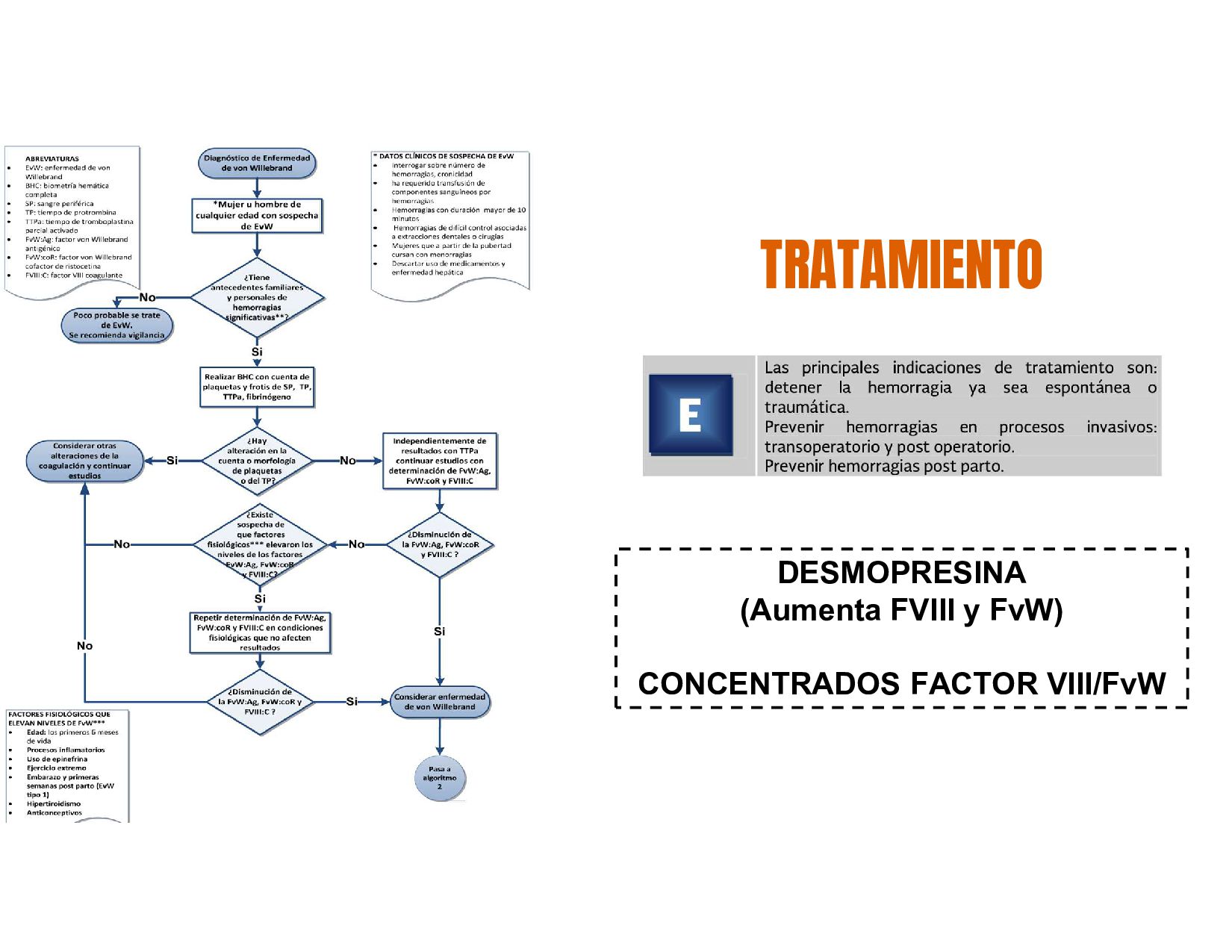
factor de von Willerbrand (FvW). El sitio mas común de hemorragias son las mucosas y varia dependiendo el grado del defecto Padecimiento hemorragico Hereditario mas frecuente Poblacion general EvW tipo 1 CUANTITATIVO 1 Y 3 CUALITATIVO 2
VON WILLEBRAND TELANGIECTASIA HEMORRÁGICA HEREDITARIA (RENDU- OSLER-WEBER) ETIOLOGÍA Autosómica recesiva Ausencia de glucoproteína IB Autosómica recesiva Fracaso de agregación plaquetaria IIB/IIIA +Frecuente Tipo I: ad cuantitativa Tipo II: ad cualitativa Tipo III: ar mixta (más grave) Adquirida: LES, gamapatías, hipernefroma Autosómica dominante: Malformación vascular congénita (Sin soporte anatómica ni capacidad contráctil) CLÍNICA Hemorragia piel y mucosas, durante acto quirúrgico y cede a medidas locales Hemorragia piel y mucosas, durante acto quirúrgico y cede a medidas locales Hemorragia piel y mucosas, durante acto quirúrgico y cede a medidas locales Lesiones en mucosa nasal, labios, lengua, TGI, GU, traqueobronquial espontaneo o al leve contacto DIAGNÓSTICO Ausencia de adhesión plaquetaria con ristocetina aun con plasma Adhesión con ristocetina efectiva pero no con ADP, adrenalina y tromboxano Alteración con ristocetina pero corrige al administrar PFC TRATAMIENTO Concentrados plaquetarios Concentrados plaquetarios Crioprecipitado o desmopresina (aumenta liberación de FVW) ALTERACIÓN HEMOSTASIA PRIMARIA (CUALITATIVO)
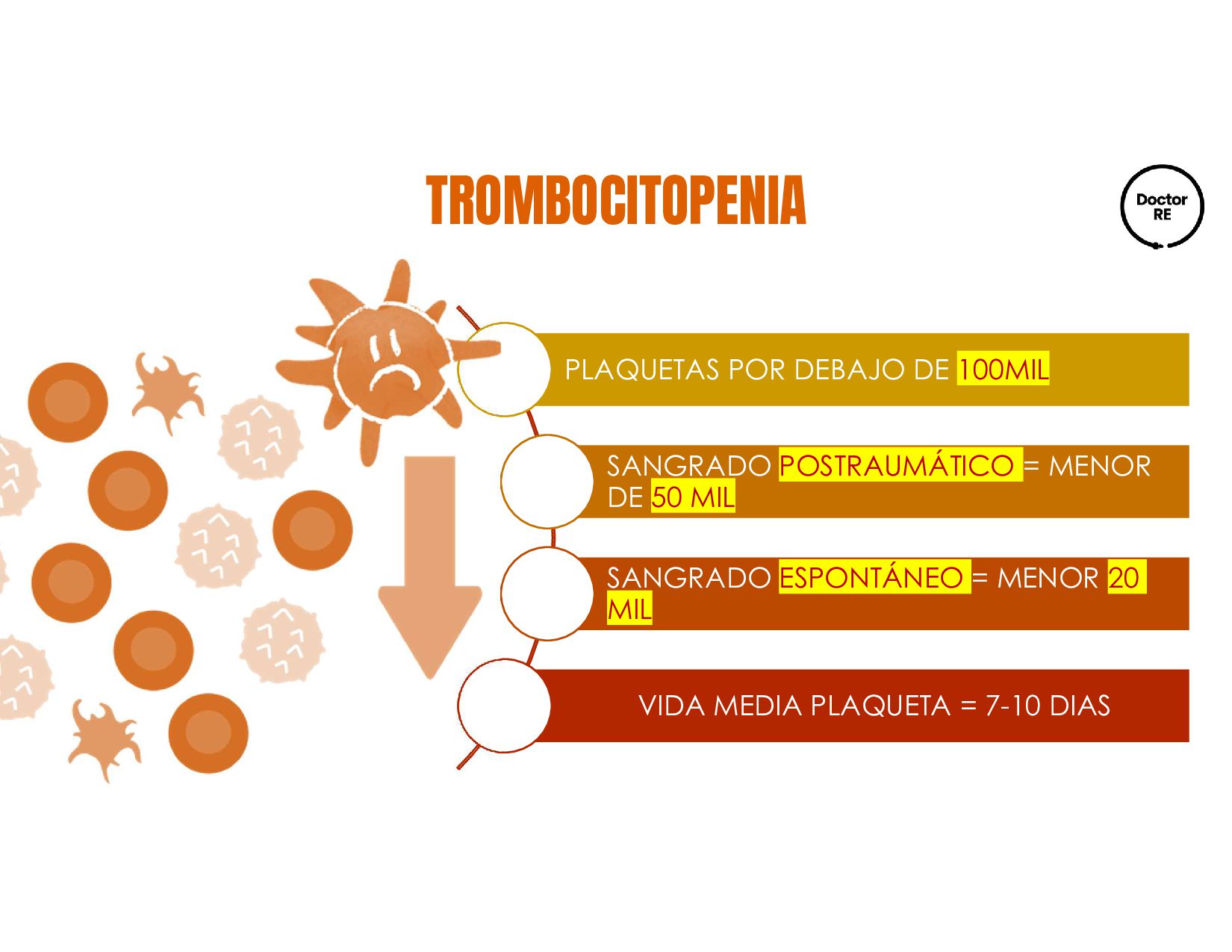
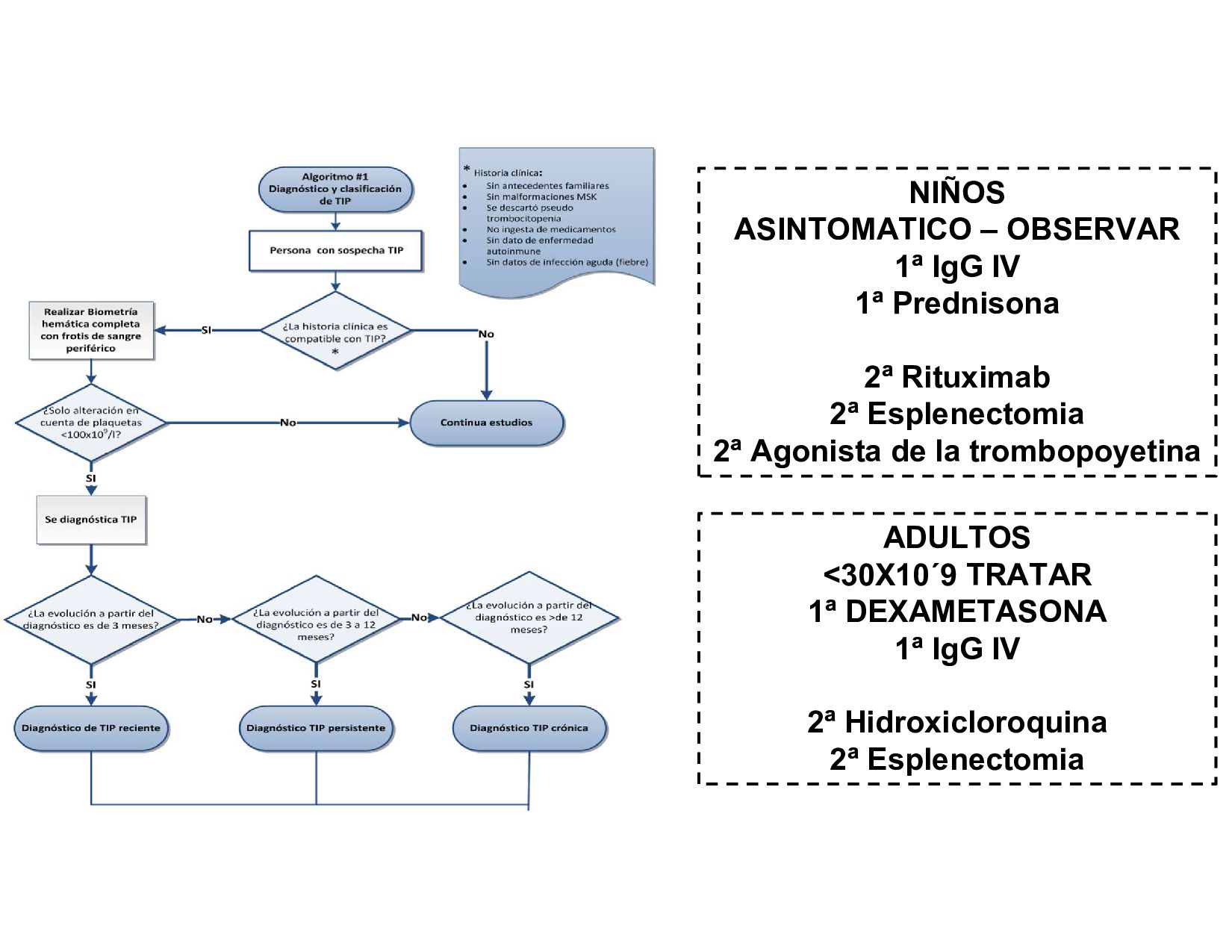
plaquetarios. Se caracteriza por conteo plaquetario <100,000/ul o 100x10´9 en ausencia de otras enfermedades o causas de trombocitopenia Casos al año Media de aparicion Mortalidad anual AUTOANTICUERPOS CONTRA GPIIB/IIIA Y GPIB 60%
genes que codifican para los factores de coagulación VIII y IX. Estos genes, se localizan en el brazo largo del cromosoma X. Varones Nacidos Vivos HEMOFILIA A FACTOR VIII HEMOFILIA B FACTOR IX MUJERES SON PORTADORAS
VON WILLERBRAND HEREDITARIA 2017 • GPC DIAGNOSTICO Y TRATAMIENTO DE TROMBOCITOPENIA INMUNE PRIMARIA • GPC DIAGNOSTICO Y TRATAMIENTO DE LA HEMOFILIA A Y B EN POBLACION MAYOR O IGUAL DE 16 AÑOS
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}