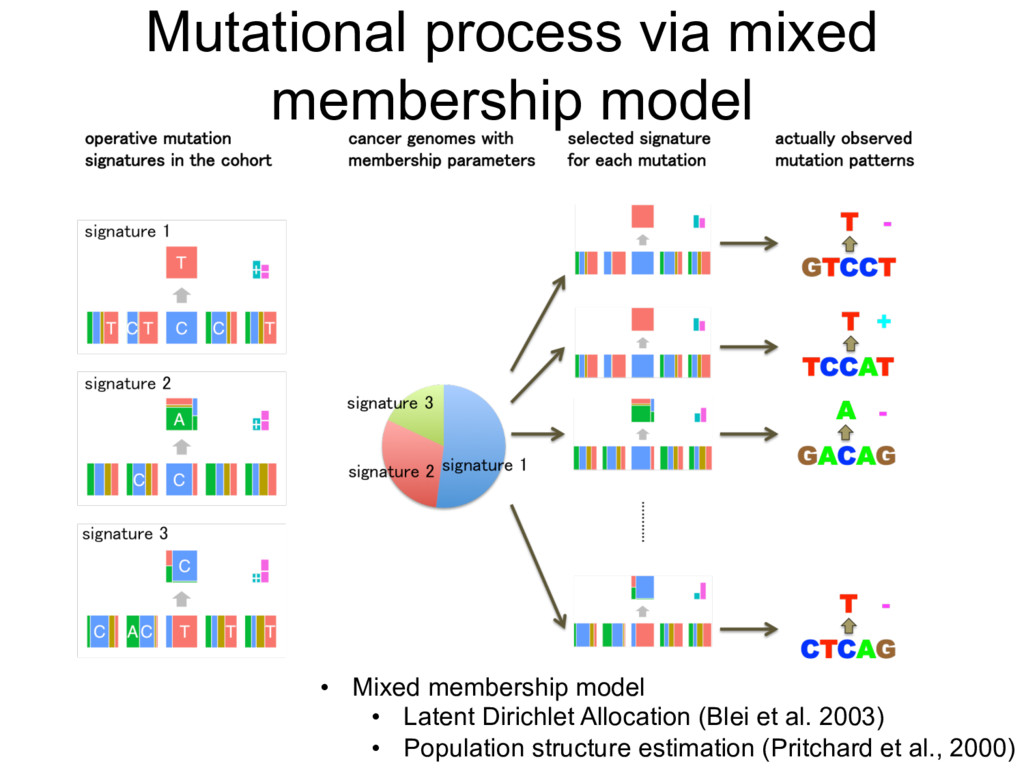
: mutation signature values for the k-th signature and the l-th features – : membership parameters for the i-th individual • EM algorithm – E-step – M-step • Used SQUARE EM (Varadhan et al., Scadinavian Journal of Statistics, 2009) o modelling mutation signatures via independent features, will grow as more and more featu orated into the analysis. ds ter Estimation meters { fk,l } and { qi } must be estimated from the available mutation data { xi,j }. Here mple approach that uses an EM-algorithm to maximise the likelihood. m denote the number of mutations in the i -th sample that have mutation feature vector m . of the EM algorithm, we calculate values of auxiliary variables ✓i,k,m defined as ✓i,k,m = qi,k QL l=1 fk,l,ml PK k0=1 qi,k0 QL l=1 fk0,l,ml . ( he M-step, we update the parameters { fk,l } and { qi,k } as fk,l,p = P m:ml=p gi,m✓i,k,m P p0 P m:ml=p0 gi,m✓i,k,m , ( qi,k = P m gi,m✓i,k,m P k0 P m gi,m✓i,k0,m . ( the R package SQUAREM [41] to accelerate convergence of this EM algorithm (SQUARE he analysis. mation and { qi } must be estimated from the available mutation data { xi,j }. Here we ch that uses an EM-algorithm to maximise the likelihood. number of mutations in the i -th sample that have mutation feature vector m . In algorithm, we calculate values of auxiliary variables ✓i,k,m defined as ✓i,k,m = qi,k QL l=1 fk,l,ml PK k0=1 qi,k0 QL l=1 fk0,l,ml . (2) we update the parameters { fk,l } and { qi,k } as fk,l,p = P m:ml=p gi,m✓i,k,m P p0 P m:ml=p0 gi,m✓i,k,m , (3) qi,k = P m gi,m✓i,k,m P k0 P m gi,m✓i,k0,m . (4) age SQUAREM [41] to accelerate convergence of this EM algorithm (SQUAREM approach to accelerate the convergence of any fixed-point iterative scheme such as Parameter Estimation The parameters { fk,l } and { qi } must be estimated from the available mu adopt a simple approach that uses an EM-algorithm to maximise the likelih Let gi,m denote the number of mutations in the i -th sample that have m the E step of the EM algorithm, we calculate values of auxiliary variables ✓ ✓i,k,m = qi,k QL l=1 fk,l,ml PK k0=1 qi,k0 QL l=1 fk0,l,ml . Then, in the M-step, we update the parameters { fk,l } and { qi,k } as fk,l,p = P m:ml=p gi,m✓i,k,m P p0 P m:ml=p0 gi,m✓i,k,m , qi,k = P m gi,m✓i,k,m P k0 P m gi,m✓i,k0,m . We use the R package SQUAREM [41] to accelerate convergence of this implements a general approach to accelerate the convergence of any fixed-p an EM algorithm). To address potential problems with convergence to loc algorithm several times (10 times in this paper) using different initial point lculate values of auxiliary variables ✓i,k,m defined as ,m = qi,k QL l=1 fk,l,ml PK k0=1 qi,k0 QL l=1 fk0,l,ml . (2) rameters { fk,l } and { qi,k } as p = P m:ml=p gi,m✓i,k,m P p0 P m:ml=p0 gi,m✓i,k,m , (3) qi,k = P m gi,m✓i,k,m P k0 P m gi,m✓i,k0,m . (4) [41] to accelerate convergence of this EM algorithm (SQUAREM lerate the convergence of any fixed-point iterative scheme such as al problems with convergence to local minima, we apply the EM his paper) using different initial points, and use the estimate with for the derivation of the above updating procedures. nto the analysis. timation fk,l } and { qi } must be estimated from the available mutation data { proach that uses an EM-algorithm to maximise the likelihood. e the number of mutations in the i -th sample that have mutation featu EM algorithm, we calculate values of auxiliary variables ✓i,k,m defined ✓i,k,m = qi,k QL l=1 fk,l,ml PK k0=1 qi,k0 QL l=1 fk0,l,ml . p, we update the parameters { fk,l } and { qi,k } as fk,l,p = P m:ml=p gi,m✓i,k,m P p0 P m:ml=p0 gi,m✓i,k,m ,
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}