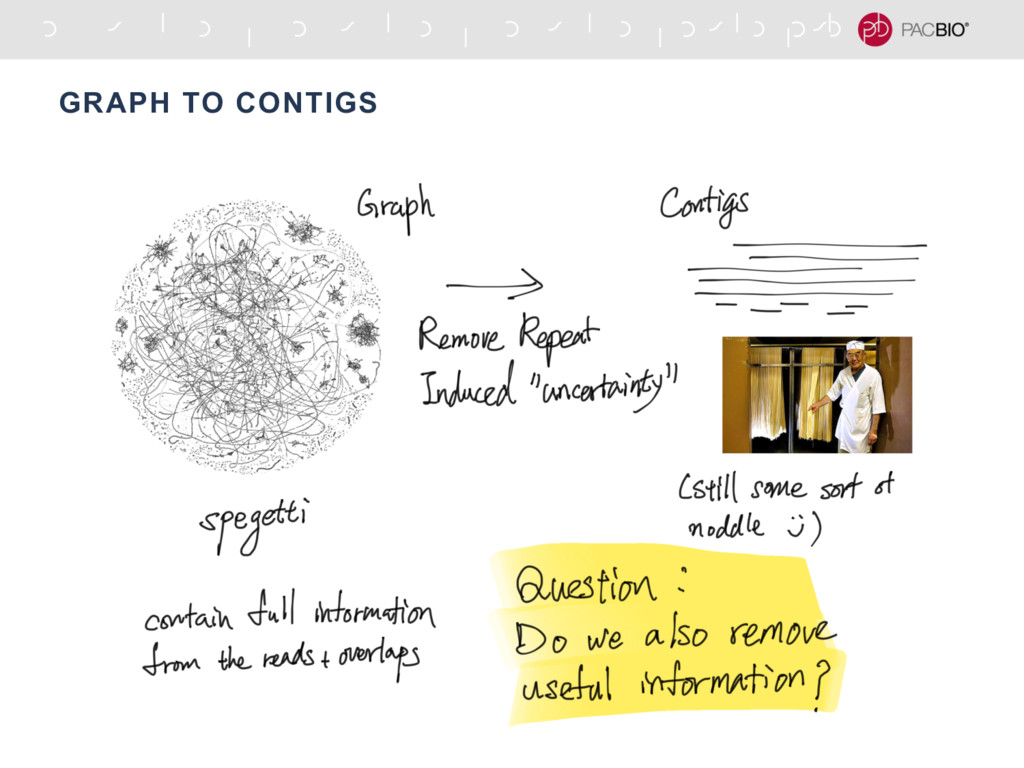
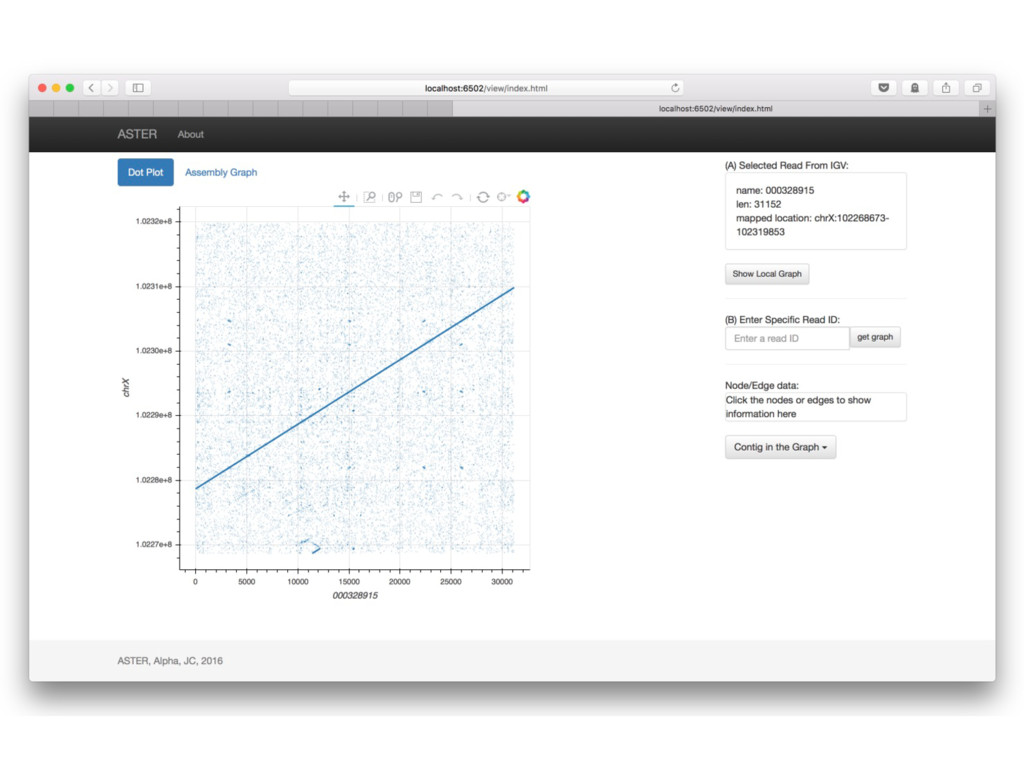
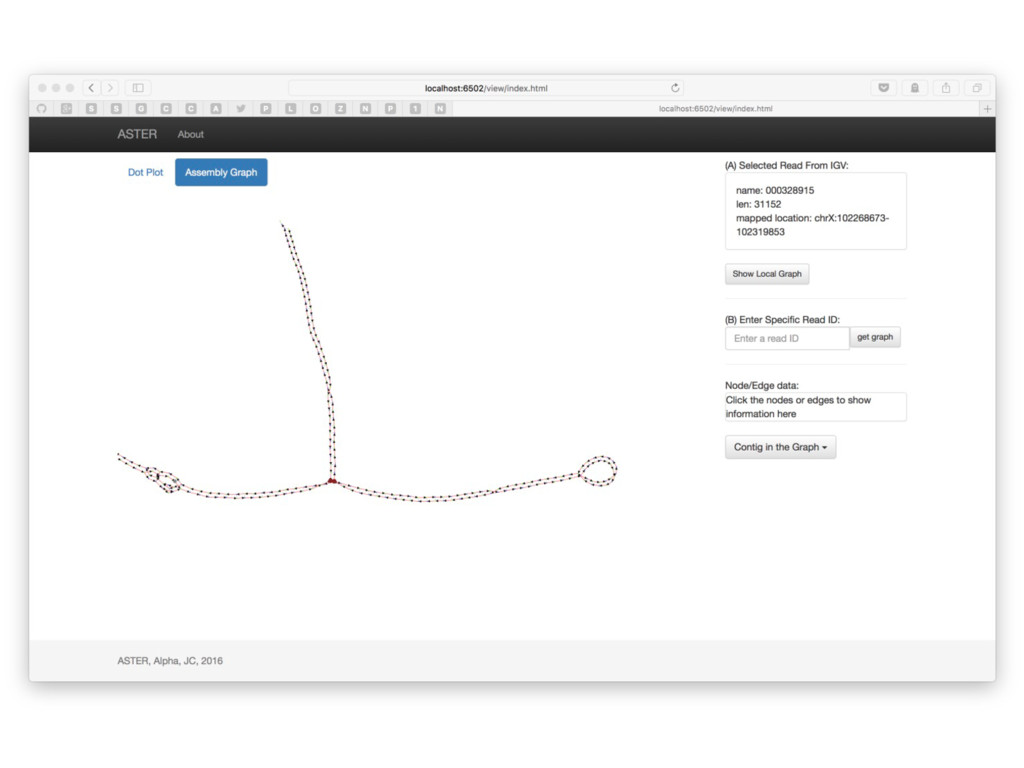
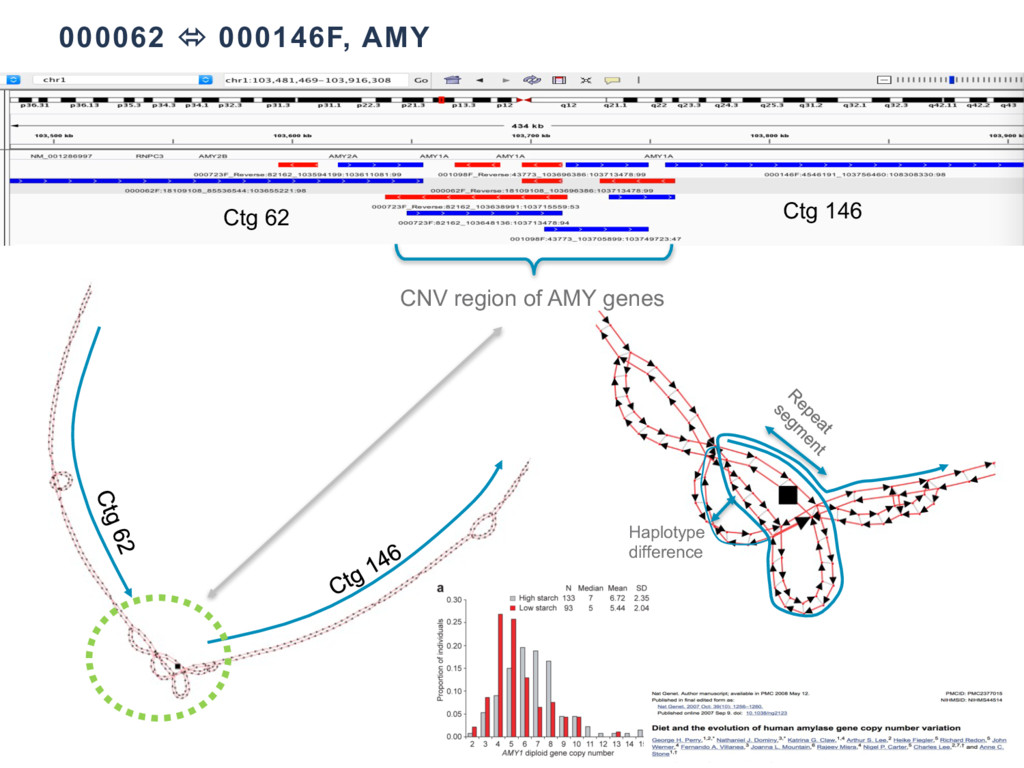
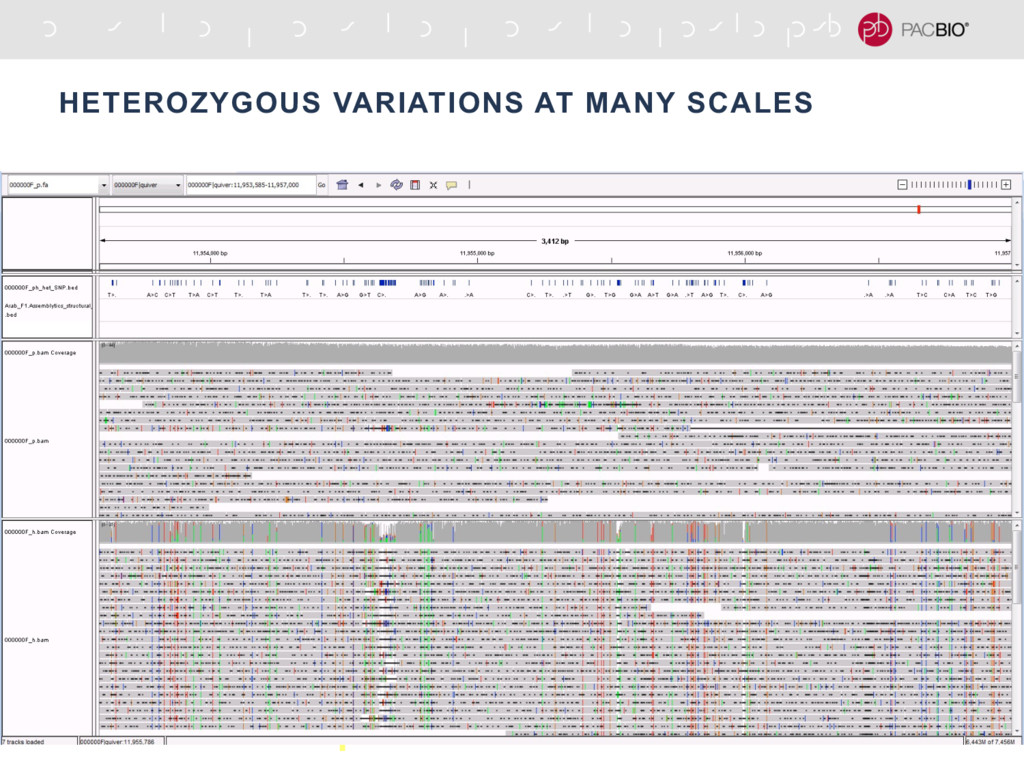
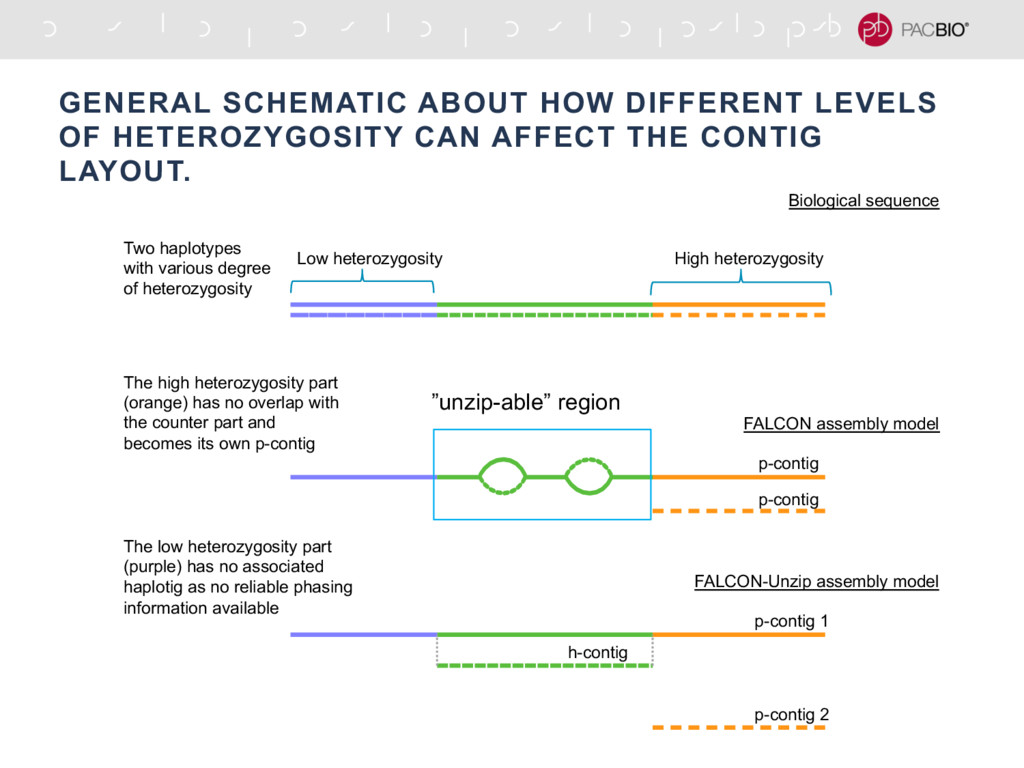
With the long reads generated with PacBio® Single Molecule Real-Time Sequencing, many algorithms developed since the late 90’s for assembling reads > 500bp have become important again. Recent advances, in aspects of both computation and algorithm, have made assembling large and difficult genomes more feasible than before. We will overview the progress from both a theoretical and a practical point of view. We compare various assembly results for human genomes with different approaches. Beyond human genomes, we discuss the complexity for assembling high heterozygosity or highly repetitive genomes and the current solutions. Furthermore, we show how to resolve repeat-induced ambiguity with detailed analysis of the assembly graph motifs and how to recover useful information lost when the assembly graph is simplified into contigs. At the end we discuss the current challenges and open problems in the ongoing development work toward generating perfect genome assemblies.
{kind=link}
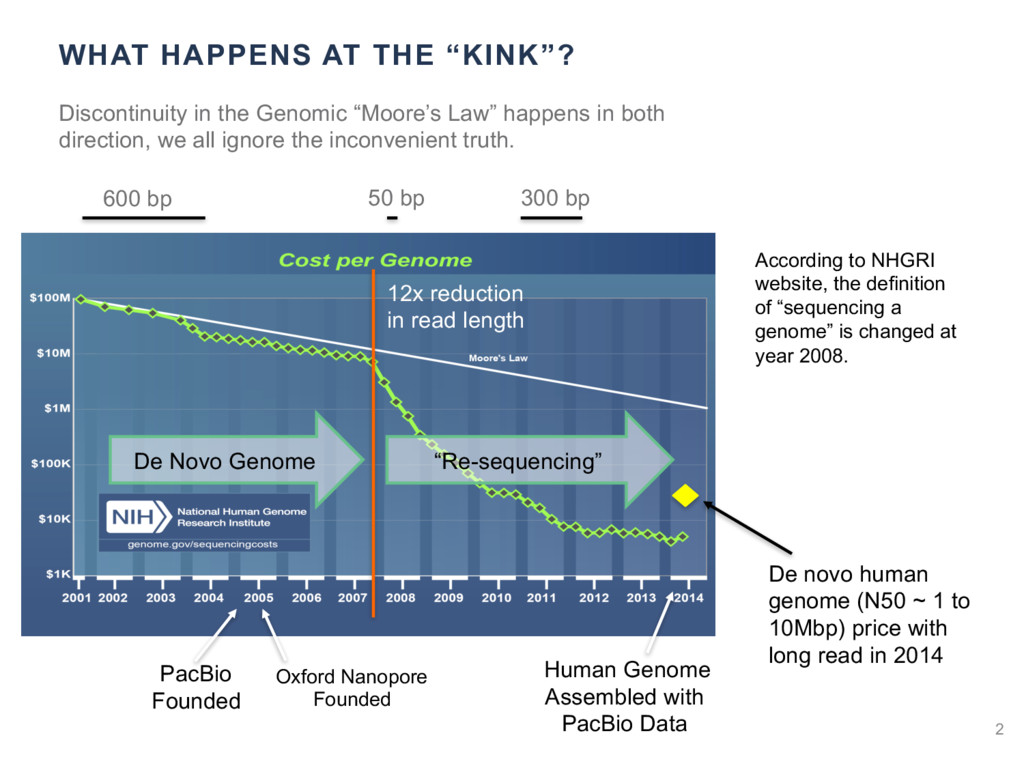{kind=link}
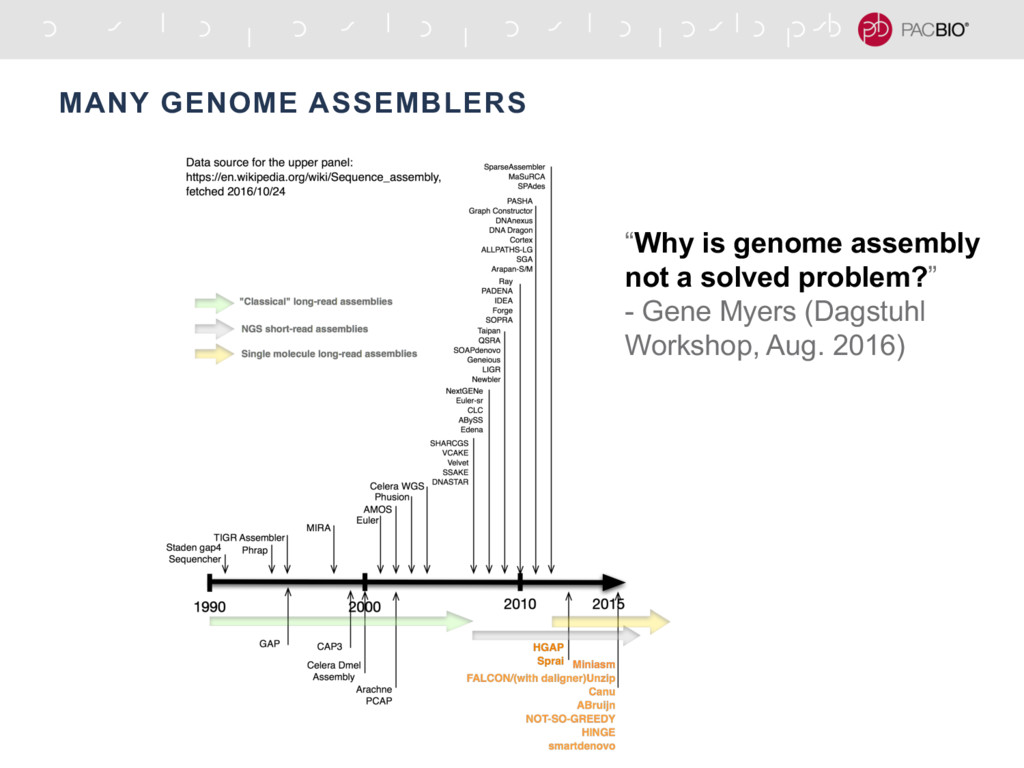{kind=link}
{kind=link}
{kind=link}
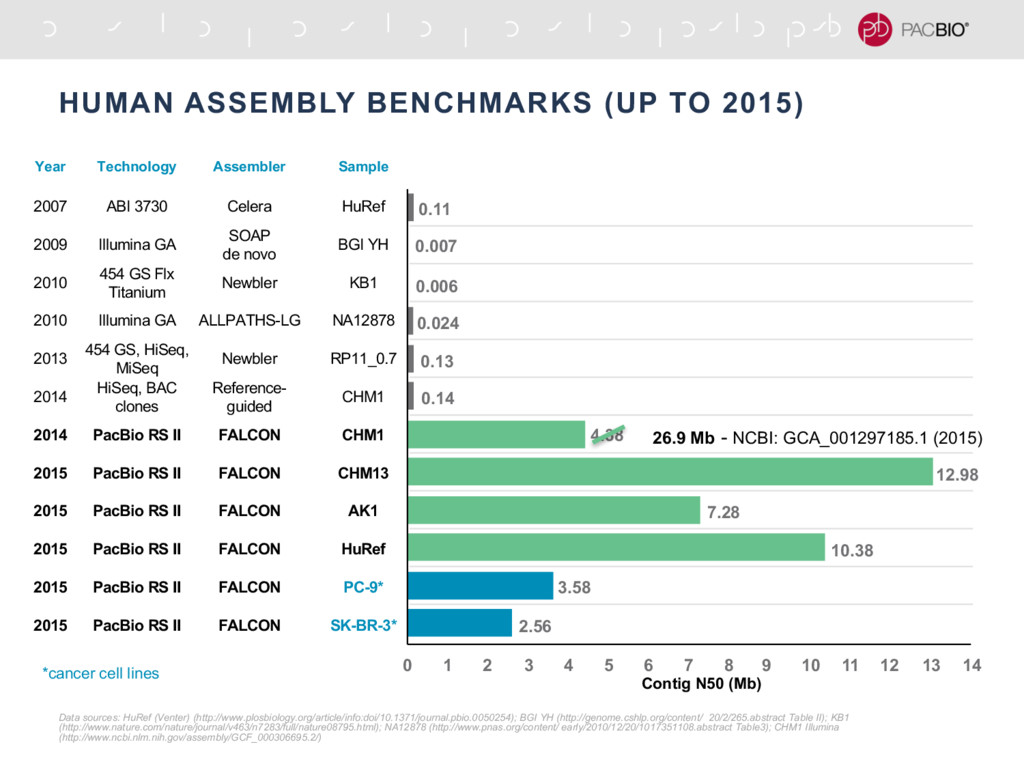{kind=link}
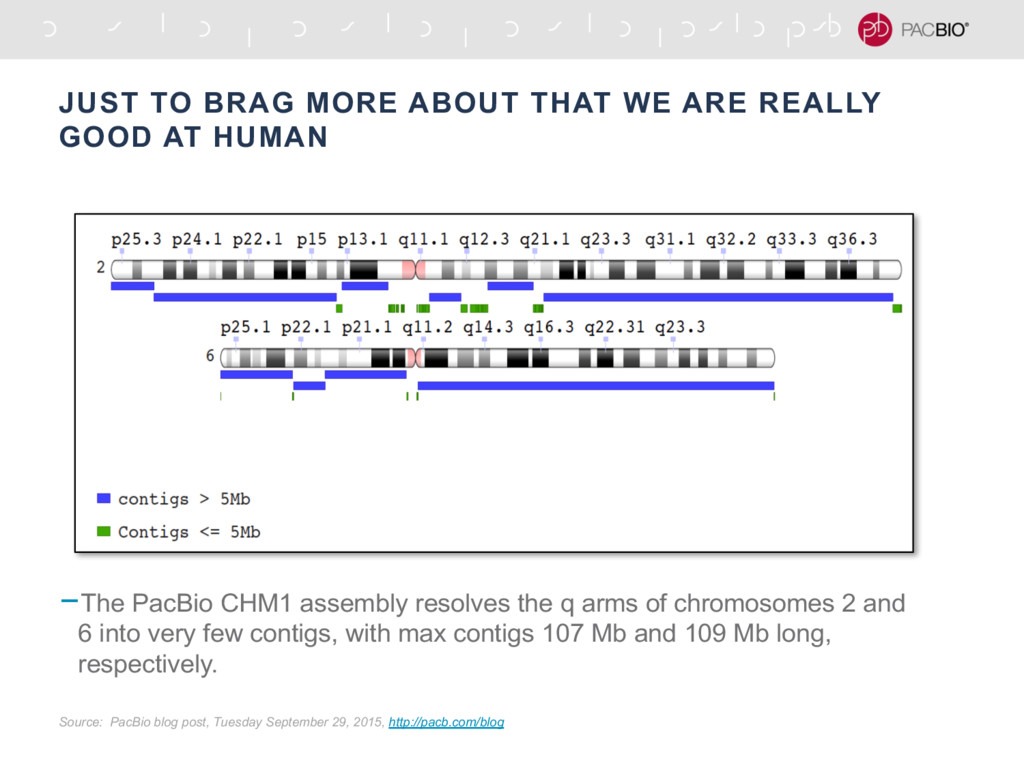{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
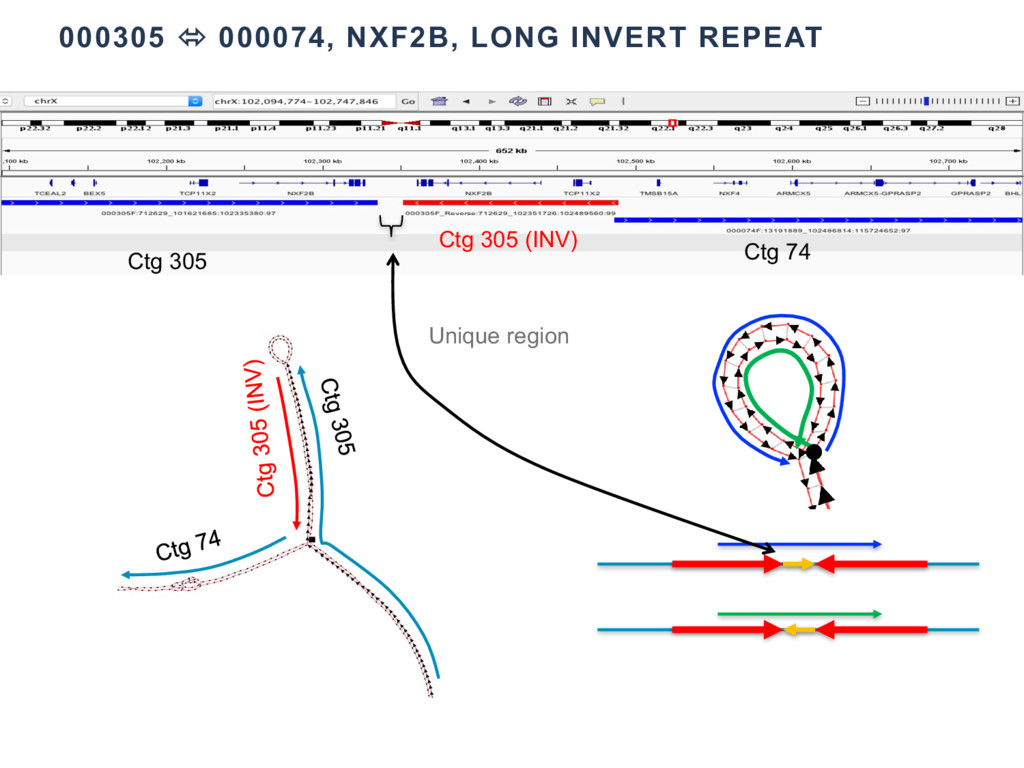{kind=link}
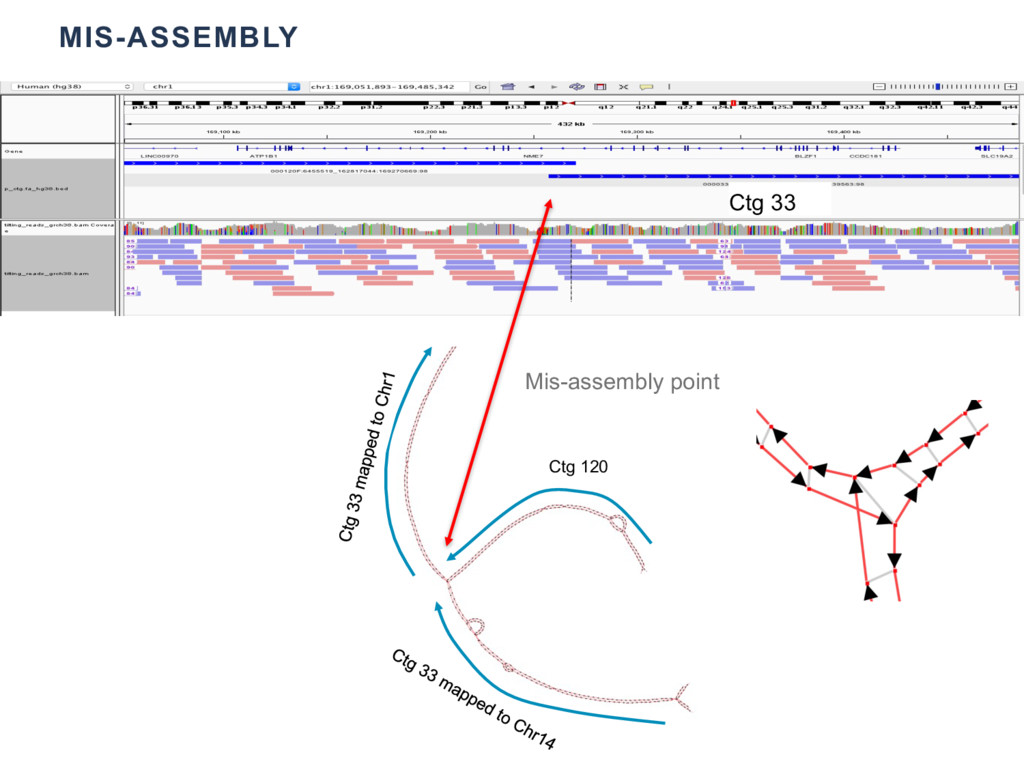{kind=link}
{kind=link}
{kind=link}
{kind=link}
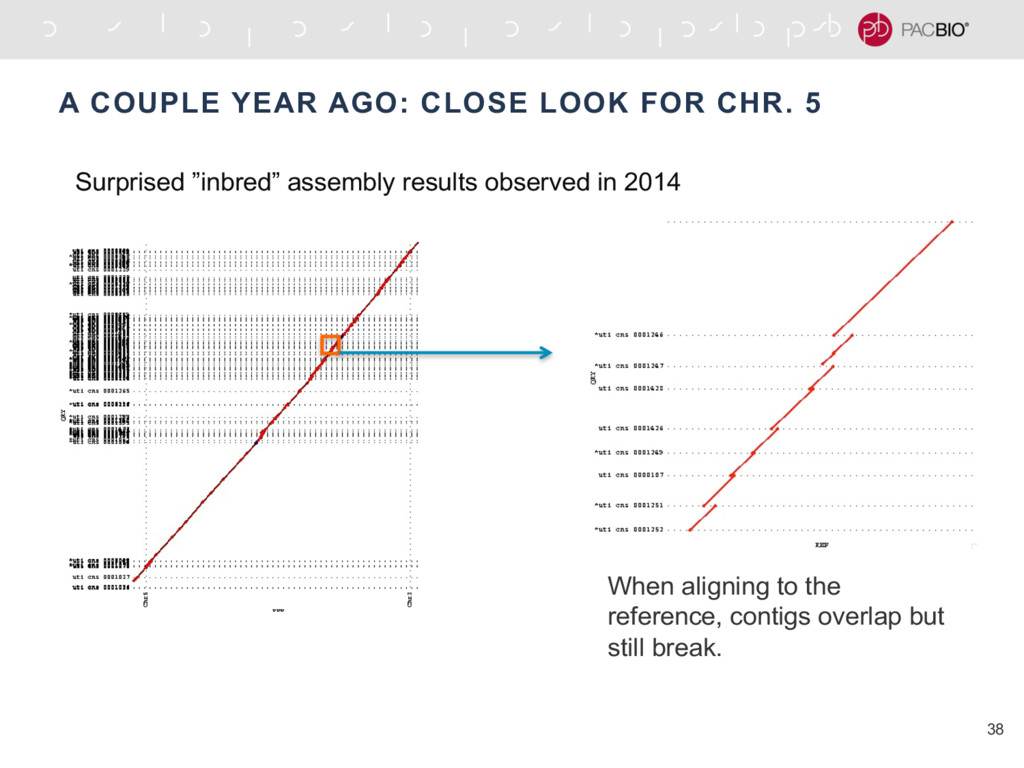{kind=link}
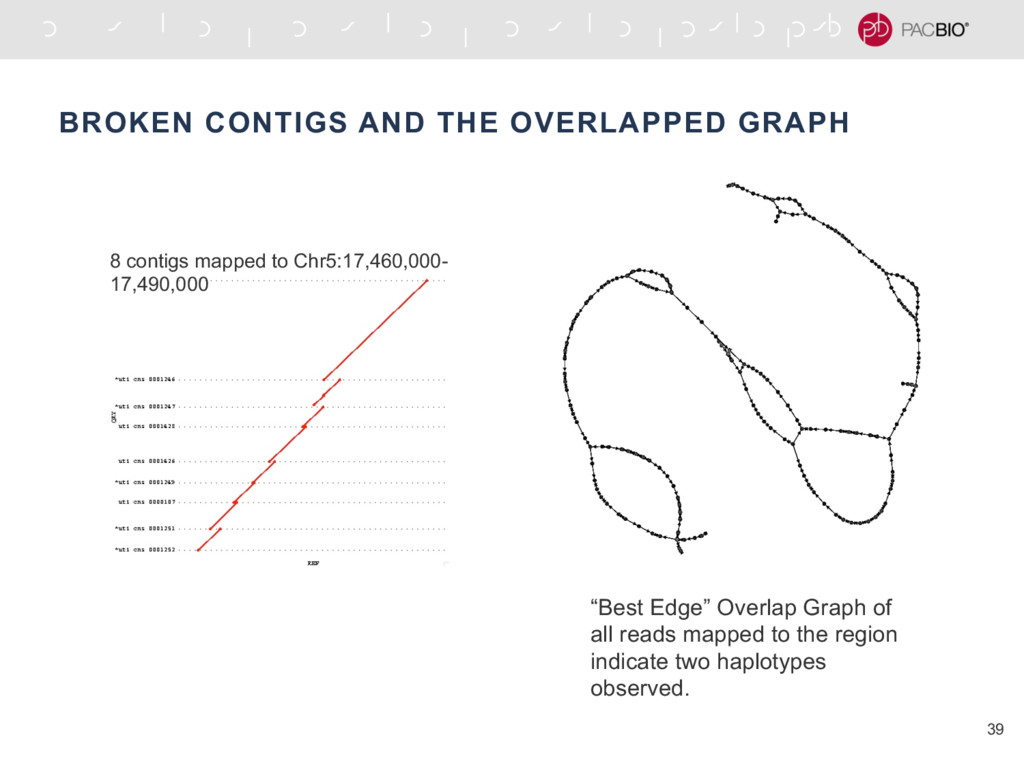{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}