How can we do super-fast genome assembly with a "SHIMMER" indexing scheme?
Abstract
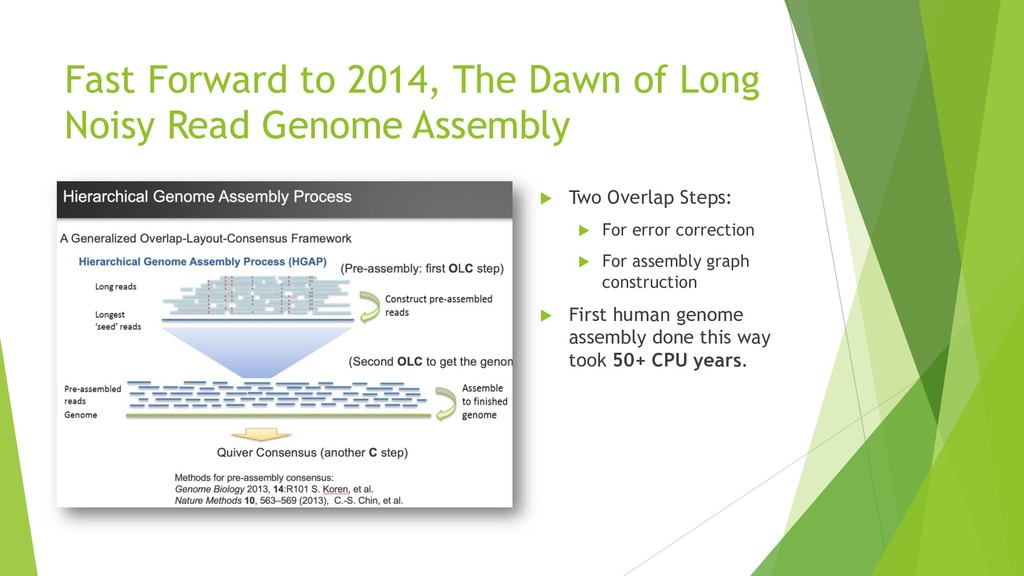
De novo genome assembly is the most unbiased way to acquire comprehensive genomic information and to gain insight for new DNA sequences that may not exist in reference genomes. Many de novo human genomes are published in the last couple of years leveraging cheaper short-read and single-molecule long-read technologies . Along with the scale of sequencing work, the computation burden persists for generating assemblies. The most common long-read assembly framework using overlap- layout-consensus paradigm requires all-to-all read comparisons. The computation complexity of this comparison step scales quadratically with the number of reads. Most methods still require hundreds to thousands of CPU hours although various techniques have been developed to reject non-overlapped pairs fast or to reduce the extra computation for repeats. High computation requirement persists for more accurate long reads (accuracy ~99% and length ~11 to 15k), which is achievable with current sequencing technologies.
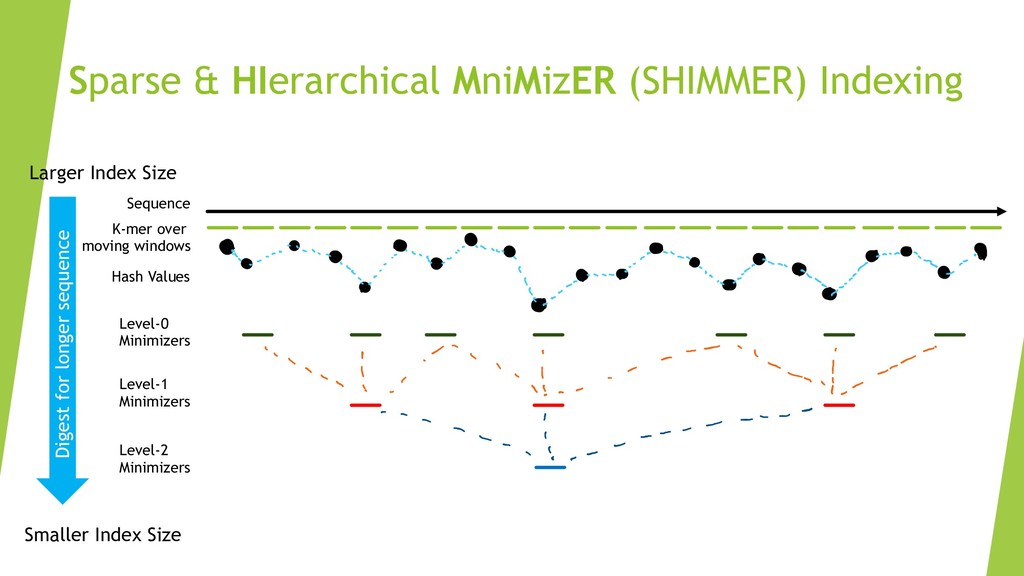
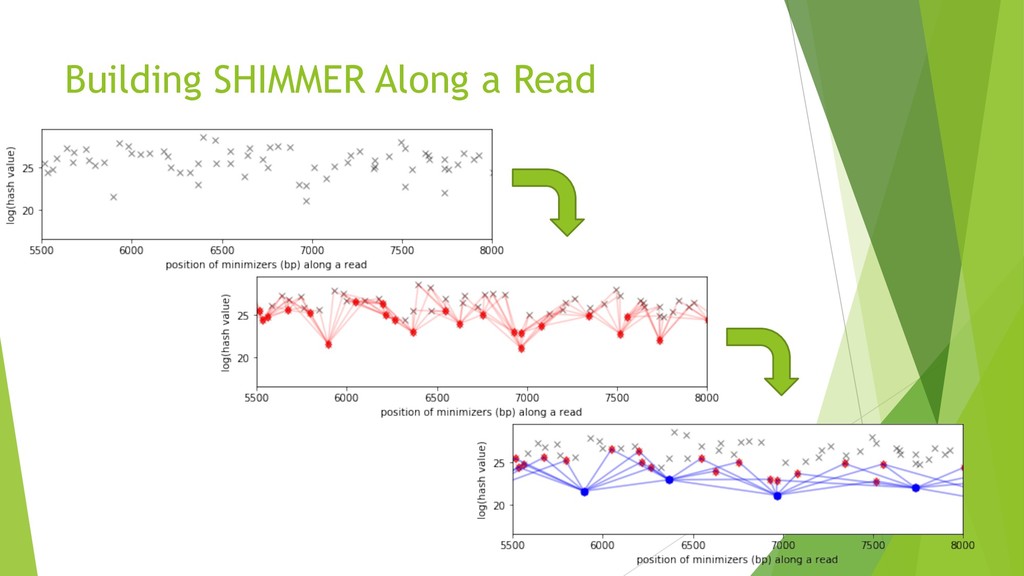
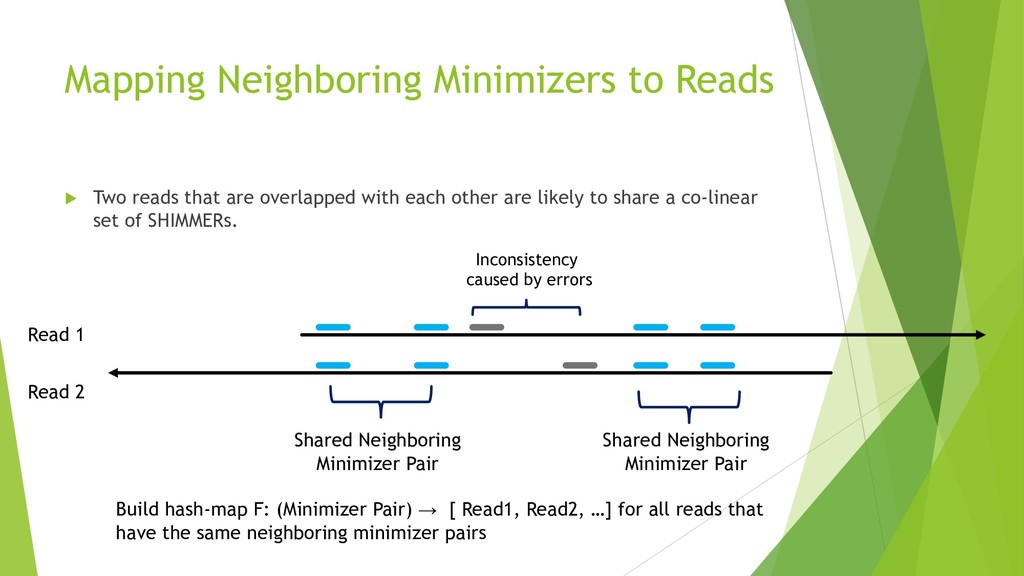
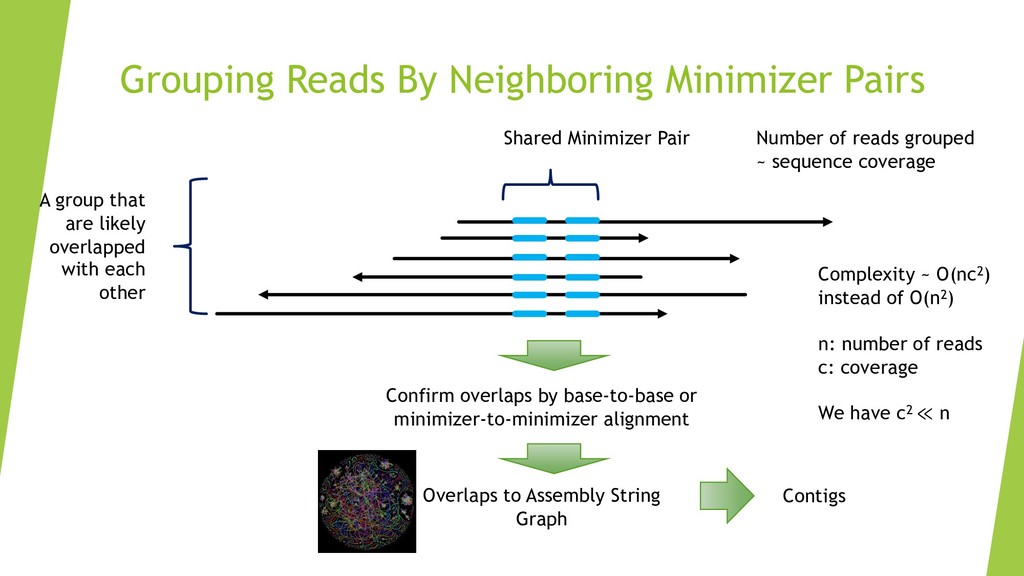
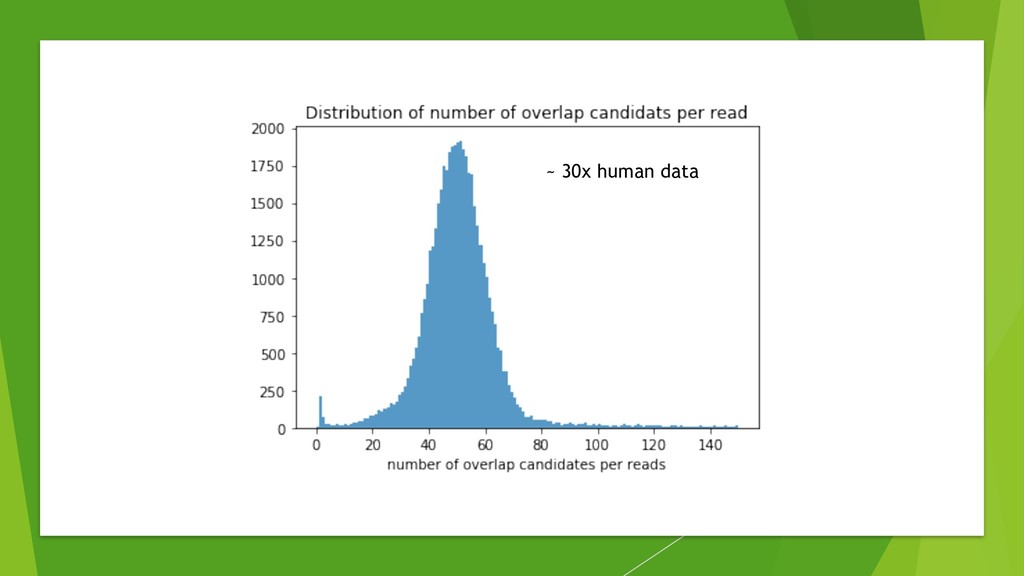
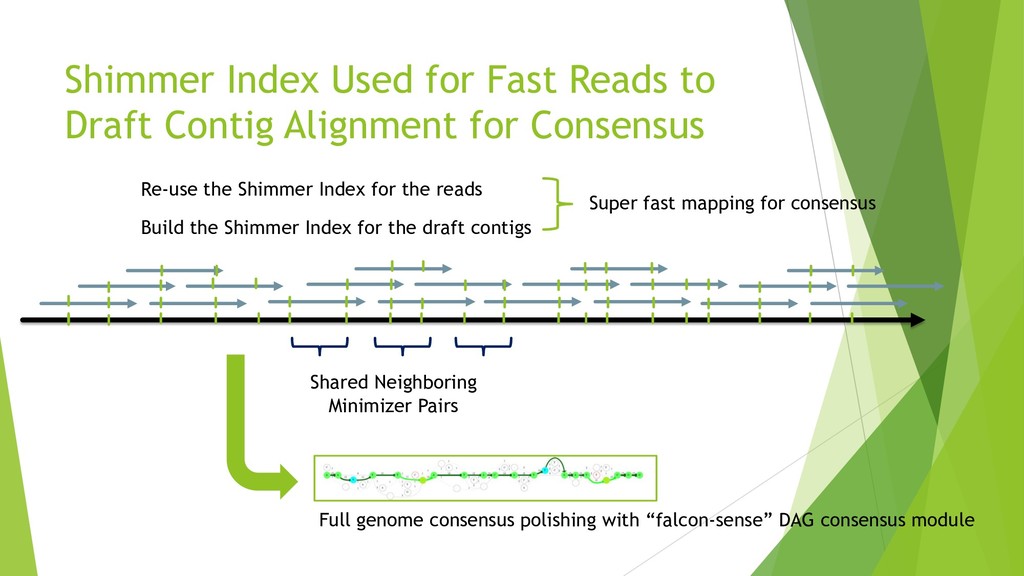
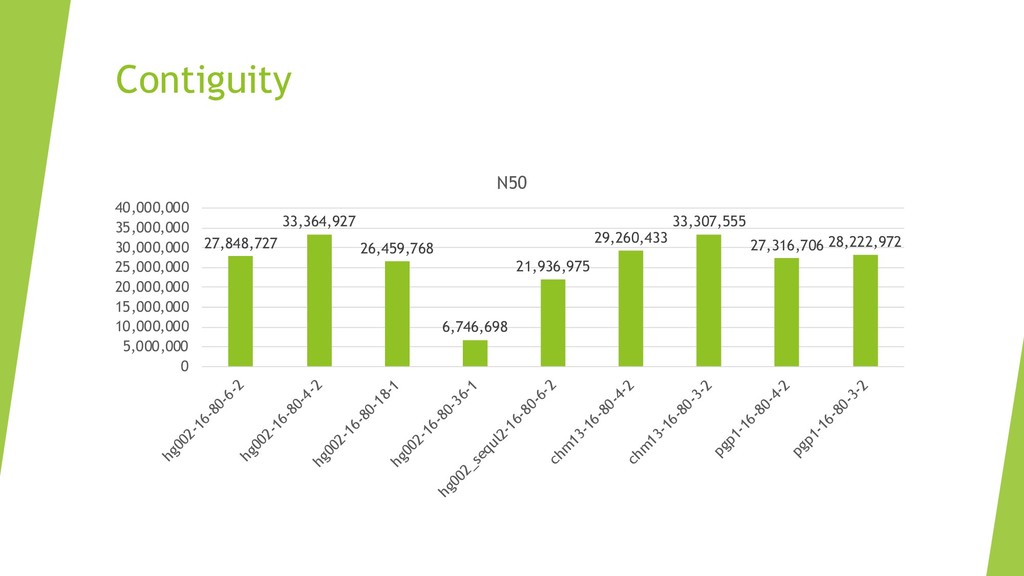
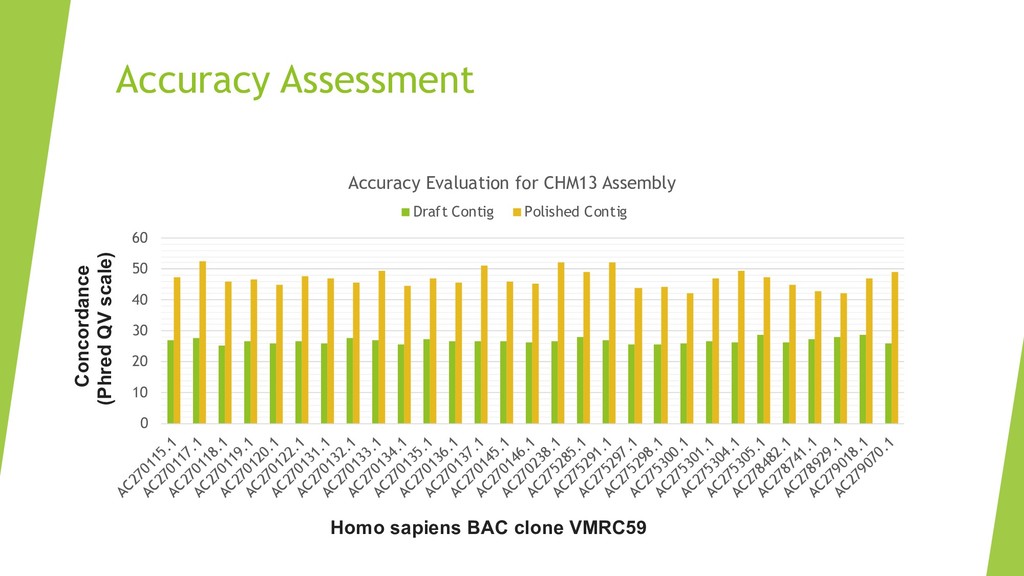
We introduce the de novo assembler Peregrine , which uses a novel minimizer based read index schema. This allows the removal of the all-to-all read comparisons. Instead, read pairs with high overlapping probability are gathered in one step and compared by utilizing the index. In our initial implementation, we can assemble 28x to 32x human PacBio CCS read datasets in less than 20 cpu hours and two wall-clock hours to high contiguity (N50 > 20Mb). The continues advent of sequencing technologies in terms of read length and based accuracy together with Peregrine will enable routine generation of human de novo assemblies. This leads to more comprehensive representation of the genomic variations on population scale beyond SNPs and small indels. We further applied Peregrine successfully to non mammalian genomes such as plants. Future implementations will enable the usage of less accurate long reads such as Oxford Nanopore and longer PacBio reads.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}