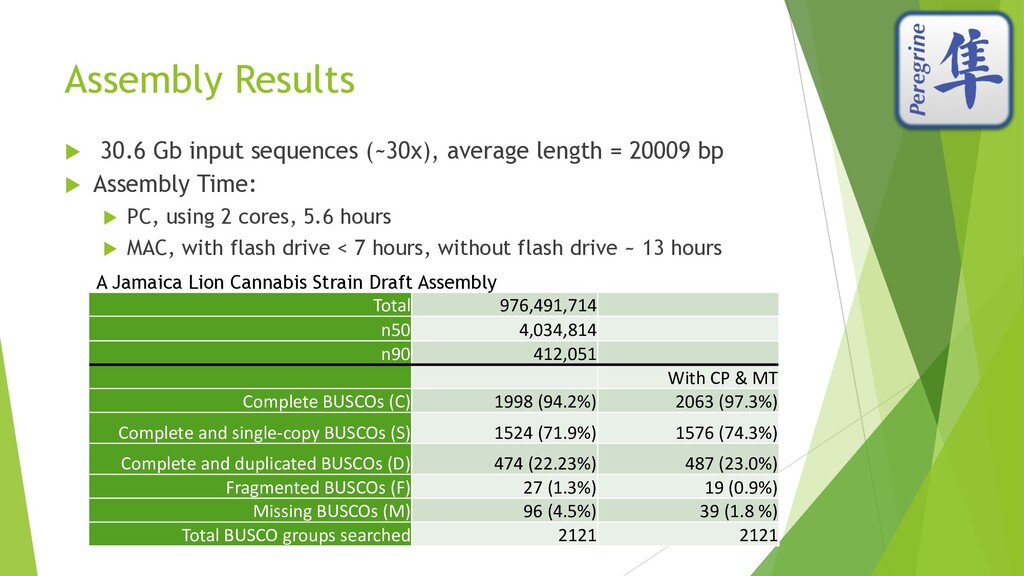
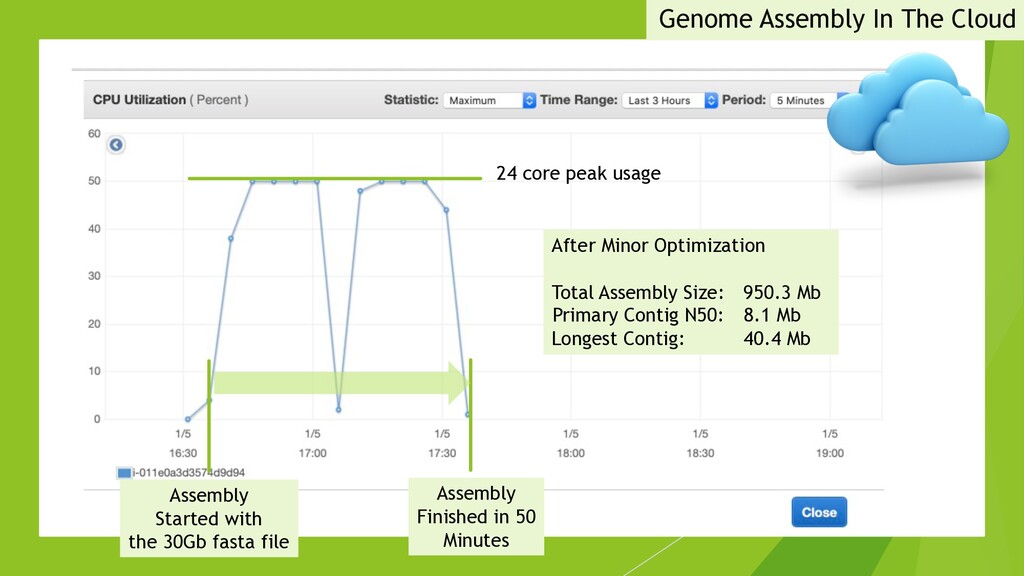
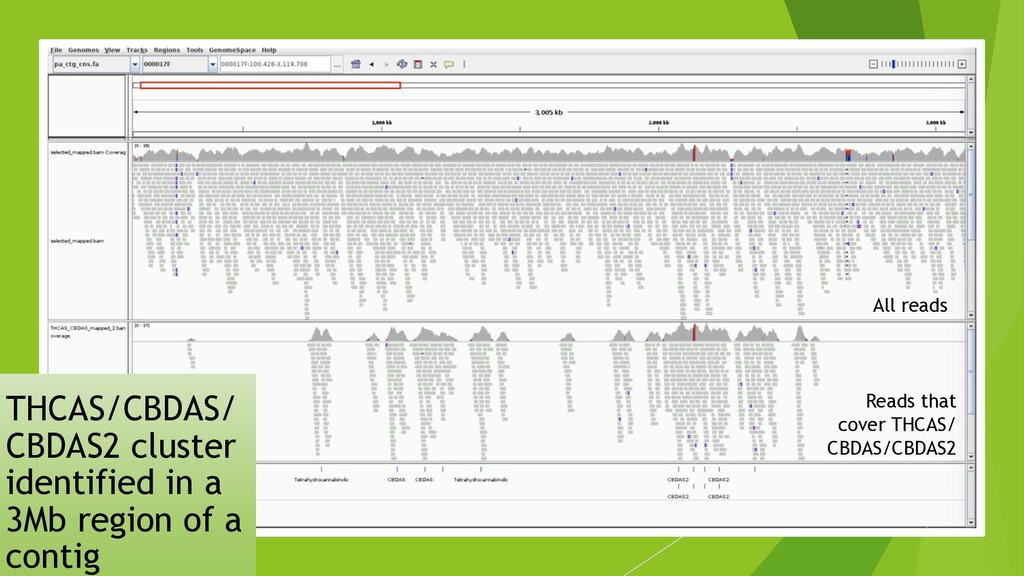
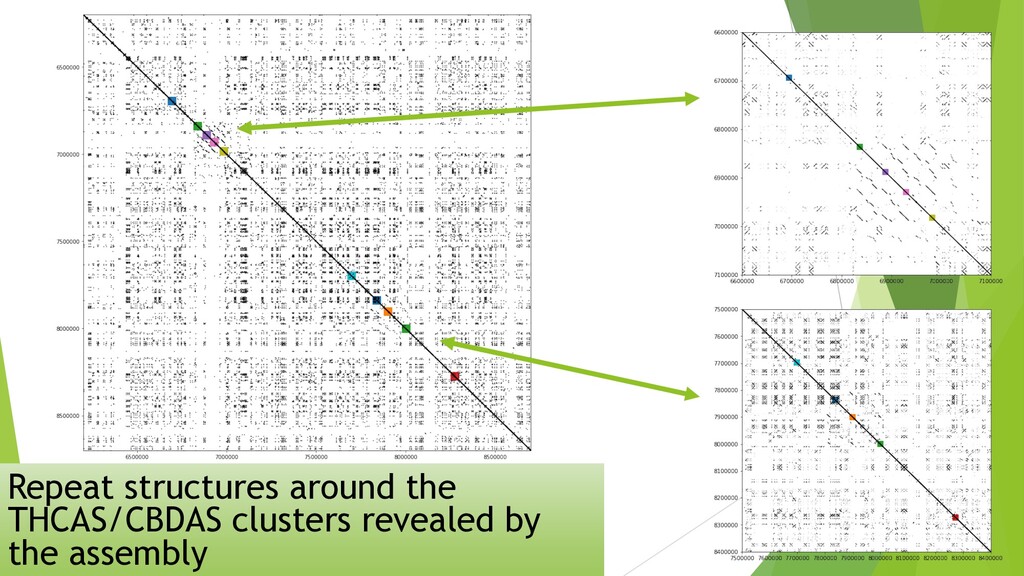
I presented this slide deck in Plant and Animal Genomics Meeting (#PAG2020 #PAGXXVIII) 2020 in San Diego. We use a novel data structure to speed up genome assembly. It becomes possible to do large genome assemblies on a home computer. In the cloud or on a server, we will be able to assembly complicated genome like Cannabis in one to two hours.
{kind=link}
{kind=link}
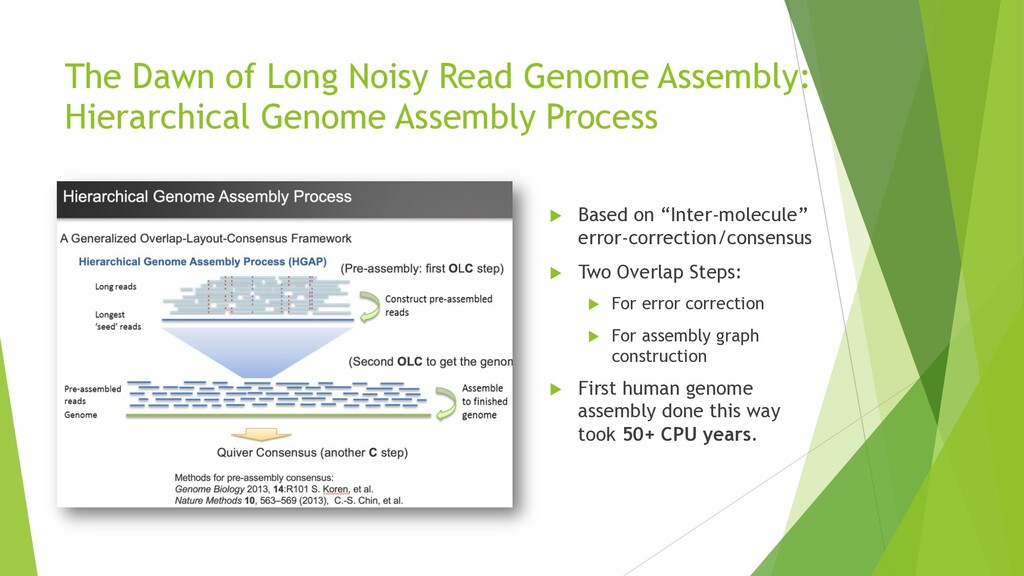{kind=link}
{kind=link}
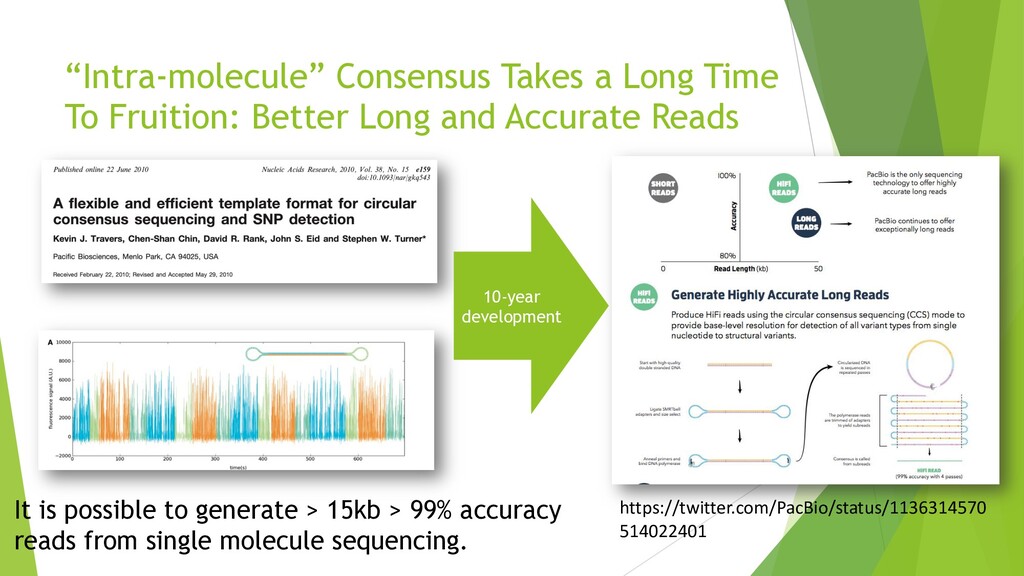{kind=link}
{kind=link}
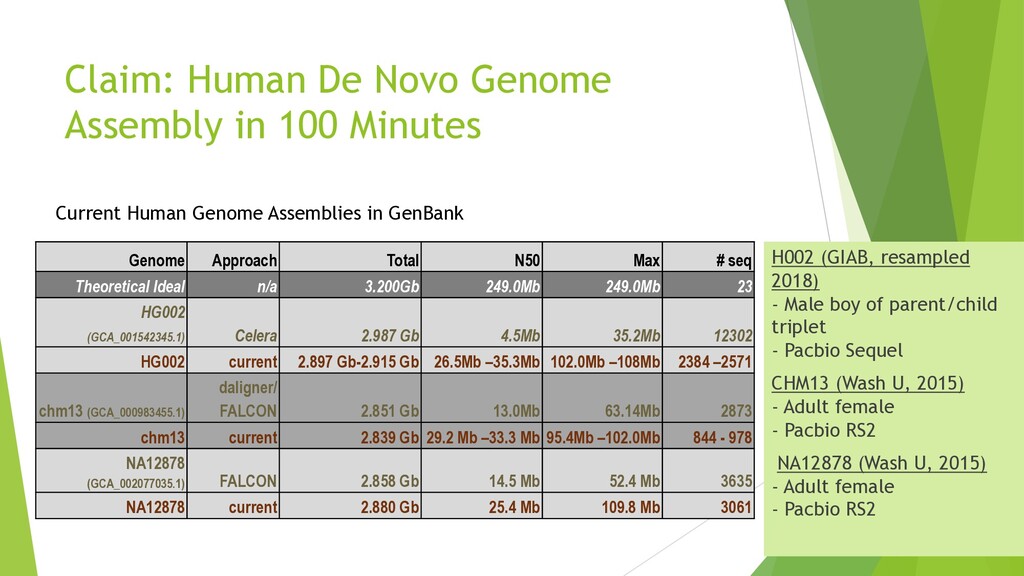{kind=link}
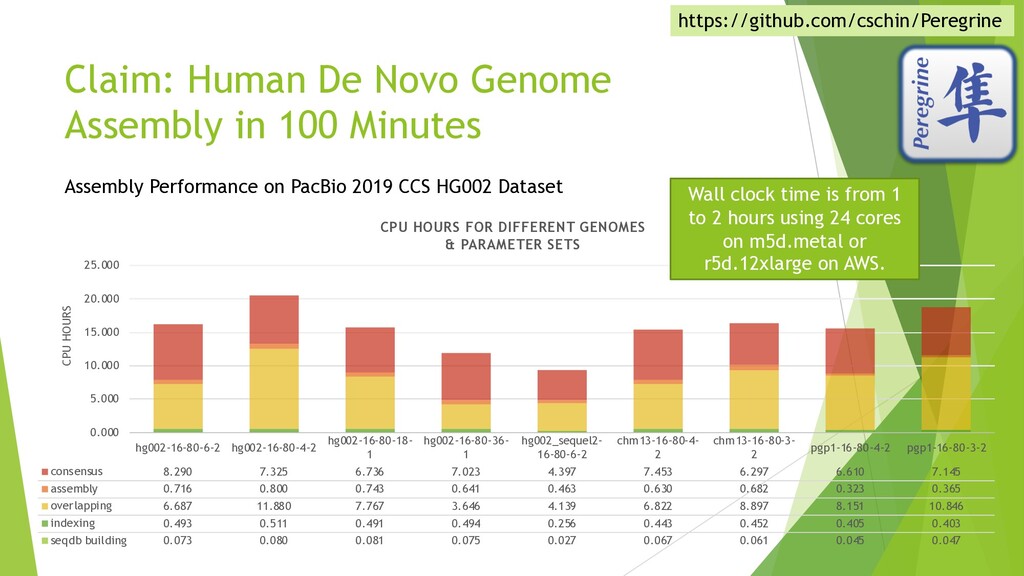{kind=link}
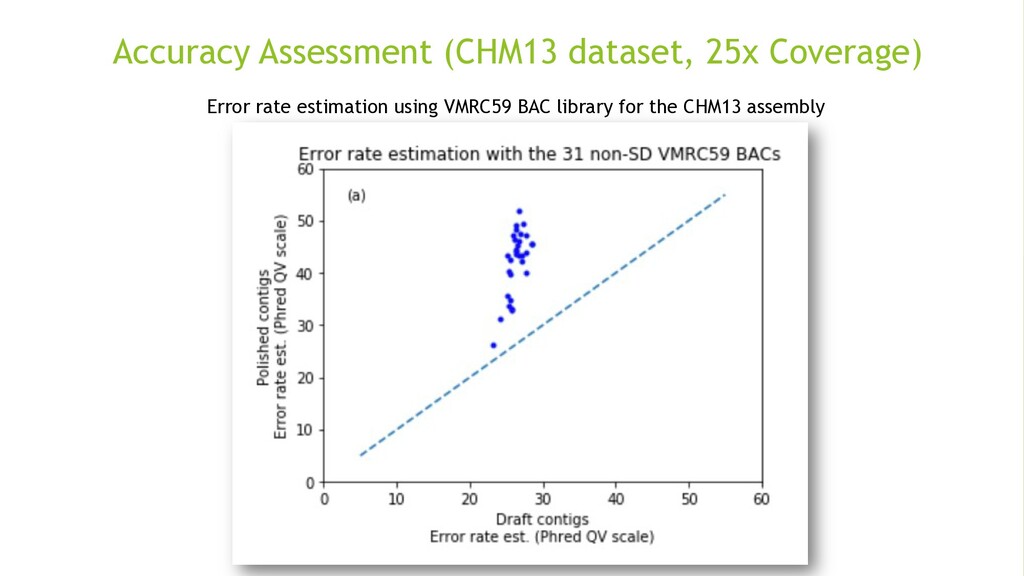{kind=link}
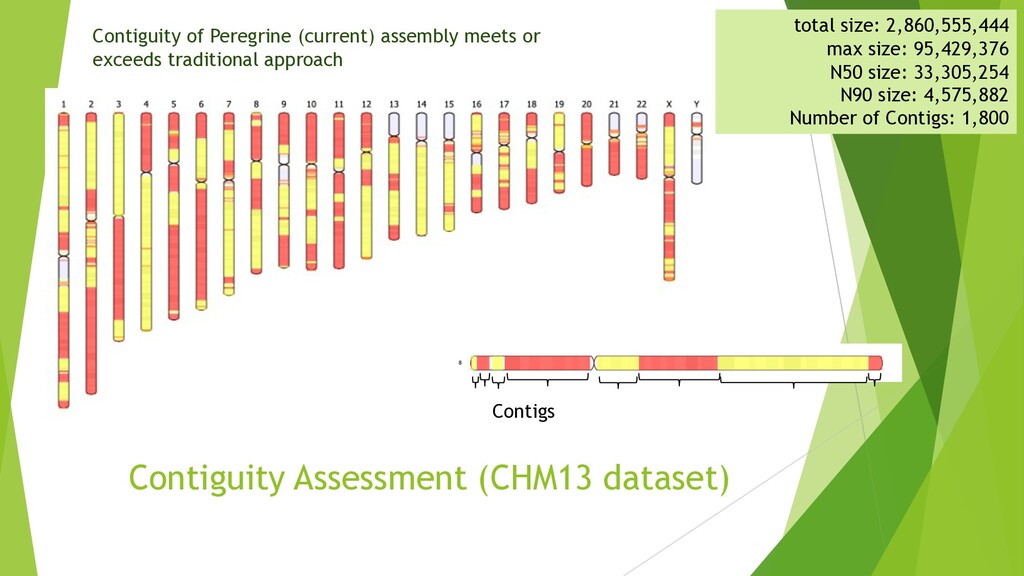{kind=link}
{kind=link}
{kind=link}
{kind=link}
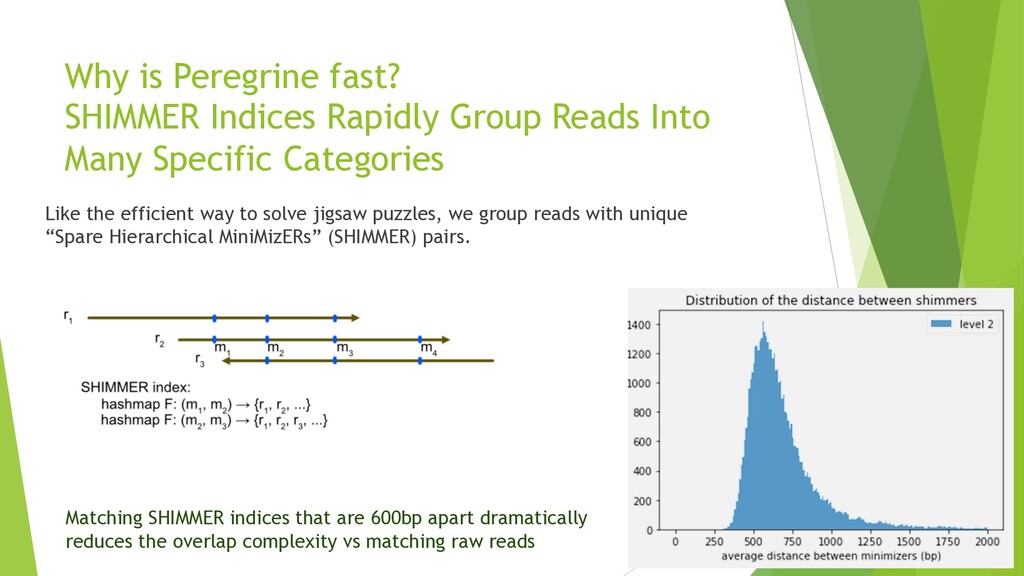{kind=link}
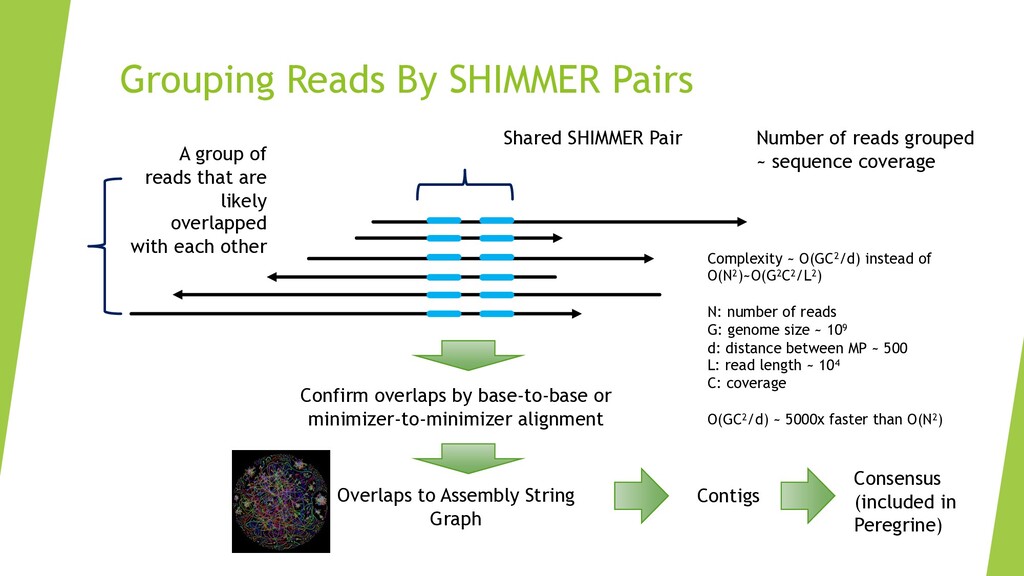{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}